Analyses of Expressed Sequence Tags from Chironomus riparius Using Pyrosequencing : Molecular Ecotoxicology Perspective
Article information
Abstract
Objectives
Chironomus riparius, a non-biting midge (Chironomidae, Diptera), is extensively used as a model organism in aquatic ecotoxicological studies, and considering the potential of C. riparius larvae as a bio-monitoring species, little is known about its genome sequences. This study reports the results of an Expressed Sequence Tags (ESTs) sequencing project conducted on C. riparius larvae using 454 pyrosequencing.
Methods
To gain a better understanding of C. riparius transcriptome, we generated ESTs database of C. ripairus using pyrosequencing method.
Results
Sequencing runs, using normalized cDNA collections from fourth instar larvae, yielded 20,020 expressed sequence tags, which were assembled into 8,565 contigs and 11,455 singletons. Sequence analysis was performed by BlastX search against the National Center for Biotechnology Information (NCBI) nucleotide (nr) and uniprot protein database. Based on the gene ontology classifications, 24% (E-value ≤1-5) of the sequences had known gene functions, 24% had unknown functions and 52% of sequences did not match any known sequences in the existing database. Sequence comparison revealed 81% of the genes have homologous genes among other insects belonging to the order Diptera providing tools for comparative genome analyses. Targeted searches using these annotations identified genes associated with essential metabolic pathways, signaling pathways, detoxification of toxic metabolites and stress response genes of ecotoxicological interest.
Conclusions
The results obtained from this study would eventually make ecotoxicogenomics possible in a truly environmentally relevant species, such as, C. riparius.
INTRODUCTION
Chironomus riparius (Chironomidae, Diptera), is widely used in aquatic exotoxicological studies for assessing acute and sub-lethal toxicities of contaminated sediments and for water monitoring due to their widespread occurrence, short life-cycle, easy to be reared in the laboratory, physiological tolerance to various environmental conditions [1,2]. To date, the endpoints used for monitoring such effects in C. riparius are based on a small number of specific biomarkers and measurements of organism level effects, such as survival and reproduction. Genomic-based techniques based on expression analysis of genes are important tools for investigating molecular level effects caused by exposure to environmental pollutants, which will provide the ability to detect mechanisms of action and subsequent adverse cellular level effects and associated with different types of toxicity [3,4]. As a pre-requisite for genomic based eco-toxicological studies knowledge of the C. riparius transcriptome is important but despite its eco-toxicological importance, no large scale transcriptome analysis of C. riparius has been done so far.
In a previous report Arvestad et al. [5] reported the transcriptome analysis of C. tentans midgut and an epithelial cell line using cDNA sequencing using conventional cDNA synthesis and sequencing method. However, with the advent of new high throughput pyrosequencing technologies using several genome sequencers, such as GS-FLX, GS-FLX-Titanium and SOLEXA, extensive cDNA sequence information can be obtained in a short period of time [6]. The GS-FLX pyrosequencer, from 454 Life Science/Roche, is well-suited for de novo transcriptome sequencing for the rapid production of sequence data with reduced time, labor and cost, and generates the longest reads [6,7]. Moreover, well characterized reference genomes of insects [8-10] could provide platforms for comparative genome analyses of non model organisms like C. riparius.
In this study, we present the first comprehensive characterization of the transcriptome of C. riparius 4th instar larvae using 454 pyrosequencing. Based on data corresponding to one single run on the FLX Gene Sequencer from 454 Life Science, almost 49,774,676 bases were assembled into transcripts and the majority of these have been annotated and functionally classified. In light of limited genomic and transcriptomic information, these data would significantly enrich the molecular aspects of C. riparius and its role in genomics based ecotoxicological studies.
MATERIALS AND METHODS
I. Insect Rearing and RNA Isolation
C. riparius strains were obtained from Korea Institute of Chemical Technology (Daejon, Korea). The larvae were reared on an artificial diet of fish flake food (Tetramin, Tetrawerke, Melle, Germany) in glass chambers containing dechlorinated tap water and acid washed sand, with aeration under a 16-8 h light-dark photoperiod at room temperature (20±1℃). The larvae were collected and total RNA samples were isolated using TRIZOL Reagent (Invitrogen Life Technology, USA). The RNA samples (A260/A280>1.8) were collected and mRNA was purified from the pooled total RNA (500 µg) by binding to oligo (dT) cellulose twice (Poly (A) Purist, Ambion).
II. cDNA Synthesis
For first strand synthesis, 10 µL of the purified mRNA (5 µg), denatured at 65℃ for 10 minutes in a RNase free tube, rapidly chilled on ice, mixed with 5 µL of 10X first-strand buffer, 5 µLof 100 mM DTT, 5 µLof dNTPs (2.5 mM each), 5 µLof Oligo dT20 (50 µM), 2.5 µLof Strata Script Reverse Transcriptase (200 U/µL) in a 50 µLreaction volume. First strand cDNA was synthesized at 42℃ for 60 minutes and cDNA synthesis and heat inactivated at 70℃ for 15 minutes and the tubes were placed on ice. For second strand cDNA synthesis, H2O (61 µL), 100 M Tris-HCl, 20 µL of second-strand buffer, dNTPs (6 µL, 10 mM each), DNA polymerase I (4 µL, 10 U/µL), and RNase H (2 µL, 1.5 U/µL) were mixed with the first strand synthesis reaction and incubated at 16℃ for 150 minutes. For end blunting, 23 µL of blunting dNTP mix, 2 µL of cloned pfu DNA polymerase (2.5 U/µL) was incubated with the second strand synthesis reaction at 16℃ for 5 minutes. The cDNA was purified using QIAquick® PCR Purification Kit (QIAGEN, CA, USA) in a final elution volume of 50 µL.
III. Library Preparation
Approximately, 1µg of the final PCR product DNA was used to generate DNA library for Genome Sequencer FLX Titanium (Roche, Mannheim, GE). The fragments ends were polished (blunted), and short two adapters were ligated onto both ends. The adapters provide priming sequences for both amplification and sequencing of the sample library fragments, as well as the "sequencing key", a short sequence of 4 nucleotides used by the system software for base calling and, following repair of any nicks in the double-stranded library, release of the unbound strand of each fragment (with 5,-Adaptor A). Finally the quality of the library of single-stranded template DNA fragments (ssDNA library) was assessed using 2100 BioAnalyzer (Agilent, Waldbronn, GE), and the library was quantitated, including a functional quantitation to determine the optimal amount of the library to use as input for emulsion-based clonal amplification.
IV. Emulsion PCR
Single "effective" copies of template species from the DNA library to be sequenced were hybridized to DNA Capture Beads. The immobilized library was then resuspended in the amplification solution, and the mixture is emulsified, followed by PCR amplification. After amplification, the DNA-carrying beads were recovered from the emulsion and enriched. The second strands of the amplification products were melted away as part of the enrichment process, leaving the amplified single-stranded DNA library bound to the beads. The sequencing primer is then annealed to the immobilized amplified DNA templates.
V. Sequencing Run, Assembly, Annotation and Functional Categorization
After amplification, the DNA-carrying beads were set into the wells of a PicoTiterPlate device (PTP) such that wells contain single DNA beads. The loaded PTP was then inserted into the Genome Sequencer FLX Instrument, and sequencing reagents were sequentially flowed over the plate. Information from all the wells of the PTP is captured simultaneously by the camera, and can be processed in real time by the onboard computer. The reads were assembled using the GS De Novo Assembler (http://www.454.com/products-solutions/analysis-tools/gs-de-novo-assembler.asp). After assembly, the resulting contigs and singlets were BlastX searched [11] against the protein databases "nr" and "Uniprot" (The UniProt Consortium, 2008). Functional categorization was done using database (http://mips.helmholtz-muenchen.de/projects/funca). The sequences were annotated using the Gene Ontology (GO) terms where possible according to molecular function, biological process and cellular component using database (http://www.geneontology.org/).
VI. KEGG Pathway Assignments
Pathway assignments according to Kyoto Encyclopedia of Genes and Genomes (KEGG) mapping was carried out using unique sequences that had BlastX scores with a cut off value of E = 10-5 The sequences were mapped to different KEGG biochemical pathways according to the EC distribution in the pathway database (http://www.genome.ad.jp/kegg/).
RESULTS
I. Sequencing, Assembly and Sequence Analysis of Pyrosequencing ESTs
To get an overview of C. riparius transcriptome, total RNA was isolated from fourth instar larvae, and mRNA purification, cDNA synthesis, and sequence determination was done. These were then pyrosequenced, and in total, 138,091 reads were obtained, constituting a total of 49,774,676 bases of cDNA. Following the assembly of the sequences, a total of 8,565 contigs and 11,455 singletons were obtained (Table 1). Among the 8,565 contigs, 3,131 and 5,434 had lengths more than 500 and 100 base pairs, respectively (Table 2).

Summary statistics of pyrosequencing of C. riparius

Length distribution of assembled contigs and singletons
After assembly, the contigs and singletons were BlastX searched [11] against the protein databases "nr" and "Uniprot" (The UniProt Consortium, 2008). Of the 8,565 C. riparius contigs, 5,102 matched proteins in "Uniprot" and 5,114 matched proteins in "nr", while the numbers for the 11,455 singlets where 4,697 and 4,710, respectively (Table 3). After removing all redundant sequences, 9,512 sequences were obtained, representing a significant part of the C. riparius transcriptome.
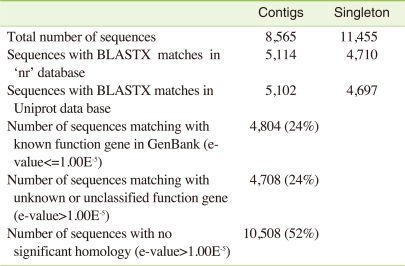
Summary of annotation of the C. riparius larval transcriptome
II. Database Searching and Functional Annotation
Gene Ontology (GO) has been widely used to characterize gene function, annotation and classification [12]. As a whole, 24% of the sequences matched with known function genes to existing gene models in BlastX searches (E-value ≤1-5), 24% showed no significant match and 52% of the pyrosequencing assemblies (E-value ≤ 1-5) did not match with any known sequences in the existing Genbank database and; thus, are likely to represent novel transcripts identified in this study (Figure 1). Similarities based on the results of BlastX searches showed the highest percentage of sequences match with Aedes aegypti (26%), followed by Culex quinquefasciatus (21%), Drosophila spp. (18%), Anopheles gambiae (15%), Tribolium castaneum (4%), Apis mellifera (2%) and Nasonia vitripennis (2%). A small number of sequences (1%) showed similarities with previously characterized genes of different Chironomus species and 6% of sequences matched with other insects and rest of the 5% matched with other (human, chicken, mouse, zebrafish, C. elegans or other organisms) species (Figure 2).

Top-ranked GO categories (molecular function) of assembled pyrosequencing ESTs.
PB: protein with binding function or cofactor requirement (structural or catalytic), SL: subcellular localization, RM: regulation of metabolism and protein function, M: metabolism, CT: cellular transport, transport facilities and transport routes, PF: protein fate (folding, modification, destination), CC: cellular communication/signal transduction mechanism, PS: protein synthesis, TC: transcription, CD: cell cycle and DNA processing, IE: interaction with the environment, IE: energy, CR: cell rescue, defense and virulence, BC: biogenesis of cellular components, SE: systemic interaction with the environment, DE: development (systemic), CF: cell fate, OD: organ differentiation, TD: tissue differentiation, GO: gene ontology, ESTs: expressed sequence tags.
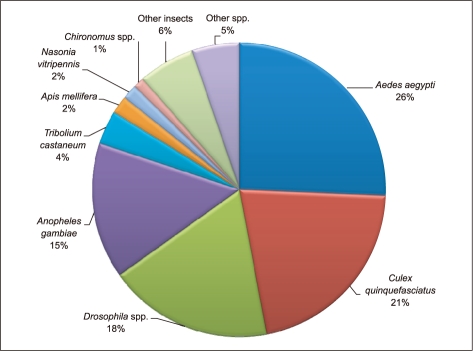
Percentage of C. riparius pyrosequence ESTs showing significant similarity with sequences in GenBank searched against NCBI.
NCBI: National Center for Biotechnology Information, EST: expressed sequence tag.
III. Pathway Analysis Based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) Classification
KEGG pathway analyses are widely used as a reference for the systematic interpretation of sequence data by linking individual genes to components of the KEGG biochemical pathways [13]. The pyrosequenced transcriptome of C. riparius was searched for the number of annotated gene sequences involved in shared specific KEGG pathways among animal phyla, using the unique sequences that had BlastX scores with a cut off value of E = 10-5. It was found that 2908 (34.73%) genes were involved in proteins with a binding function or cofactor requirement, 1,165 (13.91%) genes were involved in subcellular localizations, 915 (11%) in metabolism, 917 (10.95%) in regulation of metabolism and protein function and 700 (8.36%) in cellular transport, transport facilities and transport routes representing the largest groups with putative function, indicating the important metabolic activities in C. riparius (Table 4). These results showed the effectiveness of our transcriptome analysis of C. riparius, and indicated many of the candidate genes involved in many pathways and cellular processes.

KEGG pathway mapping for C. riparius ESTs
IV. Genes of Ecotoxicological Importance
A closer examination of the annotations revealed several genes that are of particular interest for environmental monitoring and ecological research. Several proteins catalyzing biotransformation of many xenobiotic and a number of important biomarkers for a large number of different compounds were present. Among the sequenced C. riparius transcripts, 117 different cytochrome P450 variants from 13 different families could be identified. Other interesting classes of proteins such as heat shock protein (24.1, 27, 67B2, 70, 90), genes coding for oxidative stress such as catalase, glutathione peroxidase, glutathione s-transferase, superoxide dismutase, thioredoxin reductase and several other biomarker genes such as metallothionein, vitellogenin were also present (Table 5).
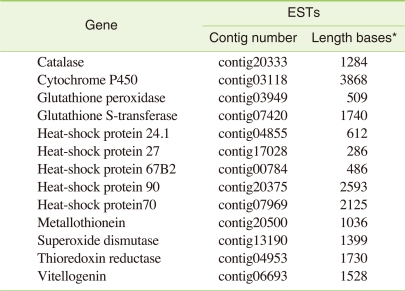
Examples of biomarker genes of ecotoxicological interest
DISCUSSION
C. riparius has been studied extensively because of their importance as an ecologically important biomonitoring species. However, due to limited knowledge of genomic resources necessary for mechanistic study, the effect of toxicants at the genomic level was rarely studied. This work describes the first assessment of the use of pyrosequencing in C. riparius and we have obtained a significant portion of the C. riparius transcriptome using this approach. To facilitate identifying sets of genes involved in a broad range of processes we developed our ESTs set from a normalized whole-body library.
As compared to Sanger-based approaches, which require cDNA cloning and bacterial transformation, transcriptome sequencing using massively parallel pyrosequencing exhibits high sensitivity and detection of low-abundant transcripts [14,15]. Transcripts that previously have been hard to sequence can therefore be detected as in the case of Arabidopsis transcriptome profiling using pyrosequecing as reported by Weber et al. [16]. Even though, the length of the sequence is shorter than as compared to Sanger sequencing, the FLX Gene Sequencer used in this study generated 3,131 contigs with an average of 924 bp length which is longer compared to previous studies [17].
In our study, sequence names to the assembled sequences were given based on the best blast match for that sequence available in the public sequence data base and almost 50% of the genes were assigned gene names. However, another 50% of the sequences not matching to known genes in public sequence databases. In our studies we obtained 9,512, non-redundant genes and thus, a major part of the transcriptome of C. riparius has been obtained. One of the limitations in non-model organisms lacking fully sequenced genome where the transcriptome pyrosequencing is based on the number of genes expressed and without a fully-sequenced genome no clear data is available.
Genome sequences for the insects D. melanogaster [8], A. gambiae [9] and A. aegypti [10] have been reported and many other species are nearing completion. In our studies 81% of the C. riparius transcriptome closely related to insects A. aegypti, D. melanogaster, C. quinquefasciatus, and A. gambiae and therefore will provide a rich source of information for further investigation using comparative genome analyses. The expression levels of unknown transcripts were also as high as those aligning in annotated regions and these transcripts are likely to represent novel transcripts, which offer possibility to study new genes which may be specific to C. riparius. In earlier reports, many unique genes are observed in transcriptome studies of M. sexta [18].
In data analysis, many genes involved in different pathways, cellular processes and genes involved in metabolism of toxic substances or well-known biomarker genes (Table 4) are identified. Since C. riparius is extensively used in ecotoxicological studies, gene expression analysis are needed for mechanistic studies to understand changes in aquatic environment and the large collection of annotated sequences produced in this study represents a reasonably complete description of the C. riparius larval transcriptome. By correlating morphological as well as physiological charecters with molecular-level responses, caused by exposure to various toxicants, the subtle effect of various toxicants could be studied.
CONCLUSIONS
Pyrosequencing offers rapid characterization of a large portion of the transcriptome and therefore provides a comprehensive tool for gene discovery. Pyrosequencing the ESTs of 4th instar C. riparius larvae resulted in the identification of many sequences of ecotoxicological relevance. Analysis of the C. riparius transcriptome has revealed several gene candidates of ecotoxicological interest and further functional characterization will identify genes with relevance to ecotoxicology. The obtained transcriptome offers the additional option to design microarrays to study transcript regulation to understand the effect of environmental stressors. Transcriptome comparison with well-studied organisms will facilitate further the understanding of how environmental stressors effect at higher organisms levels using C. riparius as a model system. The platform will allow correlation of molecular-level responses, caused by exposure to various toxicants, to the unique morphological and physiological characters of C. riparius.
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MEST) (No. 2010-0027722).
Notes
The authors have no conflict of interest to declare on this study.
This article is available from: http://e-eht.org/