Introduction
Environmental particulate air pollution is measured by a global sampling convention called particulate matter 10 (PM10) that measures the mass of particles collected with a 50% efficiency for particles with an aerodynamic diameter of 10 µm. Attention has focused on PM10 in cities because that is where most deaths occur, where pollution is routinely monitored and hence any associations are best seen. Typical urban PM10 is comprised of up to 50% by mass of combustion-derived, ultrafine carbon-centered particles with associated metals including transition metals. Other major components include ammonium salts of nitrogen, sulfur and chlorine plus geological dust and organic matter [1].
During the last decade or so, there has been growing concern about the influence of particulate air pollution on human health in urban areas. The adverse health effects caused by particles have usually been related to the mass concentration of particulate matter less than 10 µm (PM10) or 2.5 µm (PM2.5) in diameter, or to the chemical compositions of these two size fractions [2,3]. More recently, questions have been raised whether ultrafine particles ([UFP], particle diameter <0.1 µm) could be the subgroup of particles responsible for most of the observed health effects [4-7].
In PM10 this fraction is mostly composed of combustion-derived, carbon-centered particles with associated hydrocarbons and metals. Donaldson and Stone [8] suggested that progress in understanding the effects of UFP in the lungs has been achieved largely through the use of surrogate particles such as ultrafine carbon black and titanium dioxide. Using these types of particles, UFP have been shown to cause oxidative stress and pro-inflammatory effects in a number of in vivo and in vitro models. The mechanism of generation of the oxidative stress is not understood, but appears related to the large particle surface area in some way. Modulation of calcium signaling also appears to be involved in the stimulation of cytokine release by macrophages in response to UFP. Effects of PM10 are seen on cardiovascular mortality and morbidity, as well as in the lung. Although the roles of UFP in these effects are not well understood, there are plausible pathways that remain to be explored [8]. Recent studies have proposed oxidative stress-induced inflammation as one of the major pathways of PM-induced respiratory diseases [9,10]. Intracellular and extracellular production of reactive oxygen species (ROSs) can activate a number of redox-sensitive signaling cascades that could, in turn, induce inflammatory responses [9-11]. The nature and degree of oxidative stress induced by particulate air pollution can depend on the chemical reactivity of the PM and the responses that they induce in specific cell populations [12].
Atmospheric particles are a complex, heterogeneous mixture of organic and inorganic compounds that are a result of primary emissions of pollutants, secondary reactions and re-suspension of dust and other particles generated by mechanical abrasion of surfaces such as tires and roads. These varied sources of particles not only determine the subsequent composition of the particle mixtures, but may also be responsible for their toxicological effects, as has been shown in the literature. Particle-related chemical compounds implicated as health hazards include: transition metals, organic carbon, elemental carbon, and polycyclic aromatic hydrocarbons (PAHs). This evidence has been generated with in vitro models utilizing residual oil flay ash, in vitro models with "real-world" particles [13], animal experiments [14] and epidemiological studies [15,16], however, the experimental study of particle component interactions is particularly difficult due to the complexity of the mixtures [17].
Properties of UFP and their association with PM2.5 or PM10 are not well quantified. Only a few investigations concerning the chemical composition of UFP have been published. In addition to these there are numerous papers dealing with particle mass size distributions, from which it may, however, be difficult to extract any information concerning UFP. A few review articles on size distribution measurements exist.
We investigated particle mass size distributions and chemical properties in the urban area and the capacity of air pollution PM to induce cytotoxicity in human bronchial epithelial (BEAS-2B) cells.
Materials and Methods
Sampling Site and Periods
The PM sampling site in ambient air was a nearby roadside in urban air in Seoul, Korea. The distance from the sampling site to the road was 10 m and the volume of traffic per day was approximately 5,300 vehicles (gasoline 49%; diesel 20%; LPG 31%).
The sampling was performed daily during December 2007 to December 2009 except during unusual weather phenomena such as rain, snow, or Asian dust (yellow sand).
Particulate Sample Collection
To characterize the mass size distributions of water-soluble inorganics associated with urban PM, the PM samples were collected by a 10-stage Micro-Orifice Uniform Deposit Impactor (MOUDI). The PM size ranges for each stage of the MOUDI were 18 to 10, 10 to 5.6, 5.6 to 3.2, 3.2 to 1.8, 1.8 to 1.0, 1.0 to 0.56, 0.56 to 0.32, 0.32 to 0.18, 0.18 to 0.1 and 0.1 to 0.056 µm of particle equivalent aerodynamic diameter. The flow rate of the MOUDI was 30 L/min±10% and Teflon filters (pore size 2.0 µm) were used for metals analysis.
PM samples to induce cytotoxic and inflammatory responses in vitro tests were collected by a mini-volume air sampler with PM10 and PM2.5 inlets. The sampling site and periods for PM10 and PM2.5 collection were the same as in the MOUDI study. The mini-volume air samplers for PM10 and PM2.5 were used to collect 24 hours at a flow rate of 5 L/min±10% on cellulose nitrate membrane filters (Hybond ECL RPN303D; Amersham Pharmacia Biotech, Inc., Little Chalfont, UK).
All the collecting substrates were weighed before and after the sampling using a filter microbalance. Before weighing, the samples were stored in a cool and dark room and then kept for about 24 hours in the weighing room at a relative humidity of about 30 to 40%.
Chemical Analysis
Samples collected on filters by the MOUDI were extracted in 1 N nitric acid by microwave and direct analysis for metals using Atomic Absorption Spectrometry (Analyst 400; PerkinElmer Inc., Waltham, MA, USA). Metals that were quantified included cadmium (Cd), chromium (Cr), copper (Cu), iron (Fe), lead (Pb), manganese (Mn), and nickel (Ni) for each stage of the MOUDI. The metal recovery rate was 96.4%, and the limit of detection was less than 10 ng for each metal.
PAH compounds analysis in PM10 and PM2.5 was performed in an accredited chemical analysis laboratory using the United States Environmental Protection Agency method [18]. Sixteen PAH compounds were identified and measured: naphthalene, acenaphthylene, acenaphthene, fluorene, phenanthrene, anthracene, fluoranthene, pyrene, benzo(a)anthracene, chrysene, benzo(b)fluoranthene, benzo(k)fluoranthene, benzo(a)-pyrene, dibenzo(a,h)anthracene, indeno(1,2,3-cd)pyrene, and benzo(g,h,i)-perylene. Filters were subjected to a 16 hours extraction with 200 mL methylene chloride in a soxhlet apparatus. The extracts were concentrated to 3 mL by evaporation followed by nitrogen evaporation of the solvent and then transferred for analysis into1 mL vials using hexane. The soluble organic fraction was analyzed using a gas chromatograph (Hewlett-Packard 5890A; Agilant Technologies, Santa Clara, CA, USA) with a DB-5 fused silica capillary column (30 m×0.25 mm i.d.×0.25 µm film thickness), a mass selective detector (Hewlett-Packard 5972), and a computer workstation. The column temperature was set at 150℃ for 5 minutes and then was increased to 300℃ at 10℃/min where it was held for 10 minutes for the analysis. Helium was used as the carrier gas, running at 60 to 80 psi. The PAHs recovery rate was 82.6%, and the limits of detection for 16 PAH compounds were ranged from 1 to 5 ng.
Cell Toxicity
Preparation of Particles for Assay
To obtain extractable organics matter (EOM) from PM10 and PM2.5, the samples were extracted using dichloromethane in an ultrasonic bath, then the absolute EOM were dried and weighted. The EOM samples were kept at -80℃ until in vitro assay. Prior to use, the EOM samples from PM10 or PM2.5 were diluted with dimethylsulfoxide, then surged for 10 minutes and mixed fully.
Cell Culture
A human bronchial epithelial cell line BEAS-2B (ATCC, CRL-9609) was purchased from the Korean Cell Line Bank in 2007. BEAS-2B cells were grown in minimum essential medium eagle (MEM) supplemented with 10% heat inactivated fetal bovine serum (WelGENE Inc., Daegu, Korea). The cells were grown in a humidified incubator at 37℃ (95% room air, 5% CO2). The experiments used triplicate wells for each treatment level and either six or nine wells per culture plate were allocated as controls. Positive controls were included to monitor changes in the BEAS-2B cell response. All experiments were replicated with at least two independent cell passages.
Cell Viability Assay
The detection sensitivity using cell counting kit-8 assay ([CCK-8], Dojindo, Kumamoto, Japan) is higher than that using other tetrazolium salts such as 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide, 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide, and 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium or water soluble tetrazolium salts, 2-(2-methoxy-4nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophe-nyl)-2H-tetrazolium, and monosodium salt, being nonradioactive, enables sensitive colorimetric assays for the determination of the number of viable cells in cell proliferation and cytotoxicity assays. CCK-8 is reduced by dehydrogenases in cells to give an orange colored product (formazan) that is soluble in the tissue culture medium. After treatment of the cells with EOMs from PM10 or PM2.5 as described above, the supernatant medium was replaced by 15 µL/well CCK-8 solution and incubated for 2 hours. The colored supernatants without particles were transferred into a clean 96-well plate and measured at 450 nm [19].
Determination of Cytokines
The culture supernatants obtained from the CCK-8 assay were analyzed for the release of pro-inflammatory cytokines such as interleukin-6 (IL-6) and interleukin-8 (IL-8). Quantitative determination of the cytokines IL-6 and IL-8 in the culture medium from exposed BEAS-2B cells was performed using sandwich enzyme linked immuno sorbant assay ([ELISA], OptEIA™; BD Biosciences, San Diego, CA, USA). The analyses were performed according to the manufacturer's instructions. For IL-6, we used both a commercial kit (R&D Systems, Minneapolis, MN, USA) and plates prepared with anti-human IL-6, biotin-conjugated anti-human IL-6, and avidin-horseradish peroxidase from eBioscience (San Diego, CA, USA). All IL-6 values were quantified using an R&D Systems recombinant human IL-6 standard. The IL-8 ELISA used the R&D Systems duo set IL-8 development kit antibodies and standard.
Measurement of Glutathione Levels
Levels of intracellular glutathione (GSH) equivalents were determined using a colorimetric detection assay. Cells were scraped and collected by centrifugation. Each cell pellet was resuspended in 5% metaphosphoric acid (Sigma-Aldrich, St. Louis, MO, USA) and the supernatant was collected to determine total GSH concentration using a commercial kit according to the manufacturer's protocol (BioVision Research Products, Mountain View, CA, USA). The absorbance at 405 nm was read by a microplate reader. The reduced GSH concentration was obtained by subtracting the oxidized from the total concentration. A duplicate culture with the same treatments was used to determine the total protein levels.
Results
PM Levels and Size Distribution
The average monthly sampling concentrations of PM10 and PM2.5 at the sampling site on the nearby roadside were 34 to 124 µg/m3 and 20 to 80 µg/m3, respectively. The average levels of PM10 and PM2.5 were 77.7±17.9 µg/m3 and 45.6±11.0 µg/m3, respectively. The levels of PM10 and PM2.5 showed seasonal variations, with the highest in the winter and the lowest in the summer. The ratio of PM2.5 to PM10 levels varied from 41.4 to 43.0% (Figure 1).
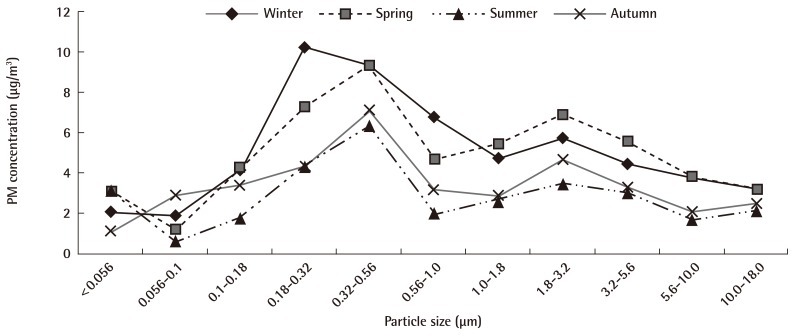
Total concentrations of all size fractions ranged from 40.16 to 109.35 µg/m3 and average concentration of PM using the MOUDI was 59.13 µg/m3. The size distribution of PM was bimodal with peaks at 0.18 to 0.32 µm and 1.8 to 3.2 µm. The percentages of UFP (<0.1 µm), fine particles (0.1 to 1.8 µm) and coarse particles (1.8 to 10.0 µm) were 7.0%, 58.5%, and 34.5%, respectively. The PM levels using to the MOUDI were showed typical seasonal variations, however, the bimodal distribution of PM sizes was similar in all seasons (Figure 2). The level of fine particles on weekends was less than on weekdays. The average volumes of traffic per day on the weekdays and weekend were approximately 6,140 vehicles and 3,060 vehicles, respectively, at the sampling site. The level of fine particles in smoggy days (PM1.0 29.2 µg/m3) was higher than on smog-free days (PM1.0 24.2 µg/m3) and the level below PM0.1 was not affected by smog (3.6 µg/m3 on smoggy day and 4.1 µg/m3 on smog-free day) (Figure 3).
PM Components
PAHs in PM10 and PM2.5
The average total concentrations of PAHs in PM10 and PM2.5 were 80.5 ng/m3 and 63.3 ng/m3, respectively, and the congener patterns were similar in PM10 and PM2.5. The major congeners of PAHs were benzo[a]pyrene, benzo[b]fluoranthene, benzo [g,h,i]pyrene, pyrene, and benzo[k]fluoranthene (Figure 4). The percentages of PAHs in PM10 and PM2.5 were 0.10% and 0.14%, respectively, the ratio of PAH levels in PM2.5 to PAH levels in PM10 was 78.6%.
Metals in Ultra fine PM
The average concentrations of Pb, Cd Cr, Cu, Fe, Mn, and Ni were 40.57, 1.40, 24.64, 82.60, 992.33, 21.29, and 25.09 µg/m3, respectively (Table 1). The mass concentrations of the metals in fine particles (0.1 to1.8 µm) accounted for 45.6 to 80.4% of the total mass concentrations of metals in PM10. The mass proportions of fine particles for pollutants related to traffic emission, Pb (80.4%), Cd (69.0%), and Cr (63.8%), were higher those for that other metals. Fe was the dominant transition metal in fine particles and accounted for 64.3% of the PM1.0 mass throughout all the samples.
Cell Toxicity
Cell Viability
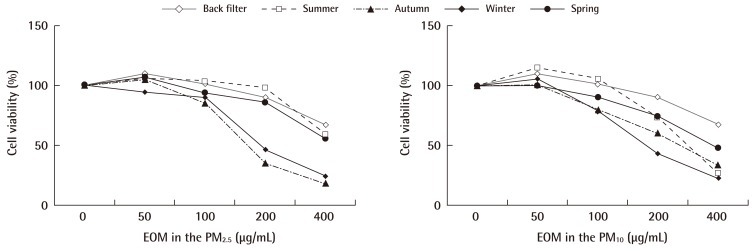
Figure 5 shows cell viability of BEAS-2B cells that were exposed to a suspension of EOM-PM for 24 hours. We observed an EOM concentration-dependent cell death rate in BEAS-2B cells for PM10 and PM2.5. Seasonal patterns of BEAS-2B cells viability (Winter<Autumn, Spring<Summer) were similar for PM10 and PM2.5. BEAS-2B cells viability decreased rapidly when exposed to more than 100 µg-EOM/mL from PM2.5.
Cytokine Release
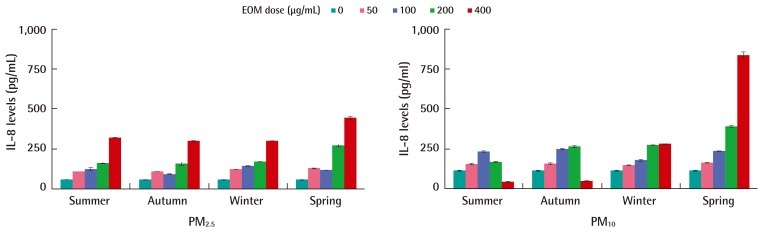
Cytokine (IL-6 and IL-8) release was dose-dependently increased in BEAS-2B cells after 24-hours exposure to EOM. IL-8 was more strongly secreted than IL-6, with a maximum of 800 pg/mL per 24 hours. Both the PM10 and PM2.5 samples showed a peak of IL-8 induction at 400 µg-EOM/mL. Cell death may limit the amount of IL-8 in the medium after 24-hour of treatment at the higher concentrations of PM10 in the summer and autumn. The IL-6 release in response to particle treatment was qualitatively similar to the IL-8 response. Although seasonal patterns of cytokine release by PM2.5 were not observed, cytokine release by PM10 in the winter and spring was higher than that in summer and autumn. Cytokine release increased rapidly after exposure to more than 200 µg-EOM/mL from PM10 or PM2.5 (Figure 6).
Oxidative Stress
Intratracheal instillation of PM10 and PM2.5 decreased GSH levels in a concentration-dependent manner. The GSH reduction ratio for PM2.5 (80%) was higher than that for PM10 (64%) and the GSH reduction ratio for PM in the winter was slightly higher than that in the summer (Figure 7).
Discussion
We measured particle mass size distributions and chemical properties in the urban area and the capacity of air pollution PM to induce cytotoxicity in human BEAS-2B cells. During the sampling period, the annual level of PM10 was less than 100 µg/m3 (the 24 hours air quality standard for PM10 in Korea), but exceeded 50 µg/m3 (the annual air quality standard for PM10 in Korea) because this site was near roadside traffic. Although we have not sampled during Asian dust storm days, the levels of PM in the spring were the most highest. There is an indirect effect of Asian dust storms in the spring period. The bimodal distribution of PM sizes in this study agrees with previous reports [20,21]. The level of PM was also related to the daily traffic volume in this study, similar to other papers [22,23].
We found weak correlations between PM mass and the concentrations of chromium, copper, lead, and iron as in the previous studies [24,25]. The ratio of PAH levels in PM2.5 to PAH levels in PM10 was 78.6% and the percentage of PHAs in PM2.5 was higher than in PM10. Due to re-suspension of dust from the ground by the wind and/or traveling vehicles, the total ambient total PAHs, as well as PM2.5, are recognized to be mainly contributed from mobile sources; those larger than PM2.5 are similarly recognized to be contributed primarily by road dust [26,27].
We showed that cell viability and cytokine release prompted by PM had seasonal variations. In particular, the higher cytokine release in the spring was considered an indirect affect of Asian dust storms in Korea, although we sampled so as to avoid Asian dust warning days. One such factor may be the content of organic compounds, which may vary in PM from different seasons. The PM contained more organic pollutants indicating incomplete combustion in the winter season and may produce more intense cell toxicity. These results also raise the hypothesis that ROSs produced from different sources may be associated with different PM-induced health effects. Several previous studies have shown that the association of PM with mortality or cardiovascular disease was stronger for the winter season [28-30]. IL-8 is one of the most potent activators and chemoattractants for neutrophils, and it contributes to neutrophilic inflammation in chronic bronchitis and emphysema [31]. It has been shown to be a potent chemotactic factor for eosinophils and T-lymphocytes as well, and therefore may have a role in the pathophysiology of allergic airway disease [32]. Fujii et al. [33] showed that primary BEAS-2B cells exposed to ambient PM produce proinflammatory mediators such as leukemia inhibitory factor, granulocyte-macrophage colony-stimulating factor, IL-6, and IL-8. The GSH system, which includes both thiol and disulfide forms of GSH along with related enzymes, provides a good example of the variation in antioxidant capacity [34]. This result agrees with previous reports that PM particles had free radical activity in vivo and in vitro, as shown by a decrease in GSH levels [34,35]. Akhtar et al. [12] suggested that PM mass concentration had an inhibitory effect on cell viability and cytokine release, and insoluble constituents of particles played a dominant role in inducing both the cytotoxic and pro-inflammatory effects. Hetland et al. [36] and Seagrave et al. [37] examined the pro-inflammatory response of cells to PM in vitro, and reported dose-response profiles similar to those reported herein.
In summary, PM size distributions exhibited a bimodal (0.18 to 0.32 and 1.8 to 3.2 µm) pattern. Fe was the dominant transition metal in fine particles and accounted for 64.3% of PM1.0 mass throughout all the samples. We found that exposure to PM2.5 and PM10 from a nearby traffic area induced significant increases in protein expression of inflammatory cytokines (IL-6 and IL-8). The cell death rate and release of cytokines in response to the PM2.5 treatment were higher than those in response to PM10.
The combined results support the hypothesis that UFP from vehicular sources can induce inflammatory responses related to environmental respiratory injury.