Introduction
DNA hybridization refers to a hydrogen bonding reaction between complementary single stranded DNA (ssDNA). It serves a basis for a number of bioassays such as microarray, membrane hybridization, and fluorescence in situ hybridization. DNA hybridization between complementary ssDNA occurs when double stranded DNA (dsDNA) denatures. DNA denaturation is a process of separating dsDNA into single strands, which are favorable to DNA hybridization. Even though the denaturation is a key reaction that determines the success of DNA hybridization based bioassays, no systematic characterization of denaturation method for dsDNA has been attempted thus far. Substantial studies have described the methods of DNA denaturation, including heating [1-3], dimethyl sulfoxide (DMSO) [4,5], and sonication [6,7]. In the above methods, the heating at high temperature (e.g., 95°C) is the most common way to denature dsDNA, particularly for polymerase chain reaction (PCR). Sonication and DMSO were also reported as boosting agents for DNA hybridization efficiency [5,7].
In this study, a series of physical denaturation (heating, heating with cold shock, beads mill with 0.1 mm beads, beads mill with 0.5 mm beads, indirect and direct probe sonication, and sonication bath) and chemical denaturation (alkaline treatment, formamide, and DMSO) were examined for the denaturation of dsDNA fragment. Well-defined dsDNA of 86 bp was used as a template of each denaturation. The degree of each denaturation was measured and the most suitable denaturation method was determined. DNA renaturation tendency was also investigated for the suggested denaturation method. The result presented in this study will provide useful data to any DNA hybridization based applications.
Materials and Methods
Preparation of DNA Materials
The DNA used for the denaturation and renaturation was part of the eaeA gene, which has been used as a common biomarker for detection of a notorious pathogen, E. coli O157:H7 [8-12]. The freeze dried E. coli O157:H7 (ATCC 43888, Manassas, VA, USA) was revived in tryptic soy broth (Difco Laboratories, Detroit, MI, USA) at 37°C (Thelco Incubator; GCA/Precision Scientific, Chicago, IL, USA) with a gentle mixing at 25 rpm (Rocker II™; Boakel Scientific, Feasterville, PA, USA) for 24 hours. The bacterial gDNA was extracted using FastDNA® SPIN Kit for Soil (MP Biomedicals, Solon, OH, USA) following the manufacturer’s instruction. Afterwards, the gDNA was used as the template for amplifying the 86-bp DNA via polymerase chain reaction (PCR). The PCR were carried out with a forward primer 5′-ACCGCGACGGTGAAAAAGAATGGG- 3′ and a reverse primer 5′-GCCCCAAGAGTTGCAGTTCCTGA- 3′, using of GoTaq® Master Mixes (Promega, Medison, WI, USA). The temperature condition for PCR amplification was 95°C for 3 minutes; followed by 35 cycles consisting of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 1 minute; plus a final cycle of 72°C for 5 minutes. PCR products were purified in a 2% agarose gel using Zymoclean™ Gel Recovery kit as well as DNA Clean & Concentrator™-5 kit (Zymo, Irvine, CA, USA) to achieve high purity. The quantity and purity of the DNA were determined using the NanoDrop™ 1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Physical Methods for DNA Denaturation
A series of simple physical methods (i.e., heating, beads mill, indirect and direct sonication) were employed for denaturing the DNA. Detailed descriptions of the four physical methods are described below.
Heating
Two variable methods (i.e., heating only and heating with cold shock) were applied to denature the DNA by Isotemp® dry bath incubator (Fisher Scientific). 10 μL of the DNA was transferred to and sealed in each centrifuge tube (VWR, Suwanee, GA, USA). For the heating only method, five groups of triplicate samples were heated at 95°C for 1, 5, 10, 20, and 30 minutes. The Final A260 of each group of sample were immediately recorded after they were removed from the incubator. For the heating with cold shock method, the samples were heated at 95°C for the same time increments, followed by being placed on ice for 1 minute, after which the A260 were measured.
Beads Mill
Two sizes (i.e., 0.1 mm and 0.5 mm in diameter) of the glass beads (Disruptor Beads™; Scientific Industries, Bohemia, NY, USA) were used for the beads mill method. 100 μL of DNA was transferred to a 1.5 mL centrifuge tube containing 50 mg of glass beads. The triplicate DNA samples were disrupted with the beads for 1, 5, 10, 20, and 30 minutes at 2,500 rpm with a Disruptor Genie bead-beater (Scientific Industries); followed by centrifuging at 3,000 rpm for 30 seconds to precipitate out the beads. The DNA recovered from the supernatant was subjected to the Final A260 measurements.
Indirect Sonication
Two ways of indirect sonication (i.e., indirect probe sonication and ultrasonic bath) were applied to the DNA. Aliquots, 50 μL of the DNA were transferred to and sealed in the centrifuge tubes. For the indirect probe sonication method, the centrifuge tube was attached on the wall of a 5 mL well, which is filled with deionized water. The probe horn was immerged in the deionized water, tilting at a 45º angle pointing at the centrifuge tube. The DNA was subject to the indirect sonication at 10 W, 22.4 kHz for 1, 5, 10, 20, and 30 minutes. The probe horn was 2 mm in diameter and connected to the XL-2000 ultrasonic dismembrator (Qsonica, Newtown, Danbury, CT, USA). For the ultrasonic bath, the bottom of the centrifuge tubes were immerged into the water of the ultrasonic bath (Branson 2510 Ultrasonic Cleaner; Branson Ultrasonics, Danbury, CT, USA) at 40 kHz for the same amount of the time. Ice was added to maintain the deionized water at room temperature during the indirect sonication treatments.
Direct Sonication
Direct probe sonication with probe horn was applied to facilitate the denaturation of the DNA. 300 μL of the triplicate DNA samples in a 1.5 mL centrifuge tube was subject to the sonication by the probe at 10 W for various time intervals (30, 60, 90, 120, 150, 200, 250, and 300 seconds). The probe was cleaned with 70% ethanol followed by deionized water for three times between the samples to prevent the contamination. The temperature was monitored with a glass laboratory thermometer after each time interval to monitor the temperature change during the sonication.
Chemical Methods for DNA Denaturation
The chemical denaturation methods looked into in this study includes a variety of concentrations of NaOH, formamide, and DMSO at ambient temperature. Detailed descriptions for the three methods used are described in the following sections.
Alkaline
Various concentrations of NaOH (0.01, 0.1, and 1 mol/L) were employed to denature the DNA fragment. The stock solution of NaOH (10 mol/L) was made with 40 g of NaOH pellets (Fisher Scientific) dissolved in 100 mL of deionized water. The NaOH was serially diluted from 1 to 0.01 mol/L with deionized water for the following experiment. For each experiment, 40 μL of NaOH (0.01, 0.1, or 1 mol/L) were added to 4 μL of the DNA in each centrifuge tube. The mixture was homogenized with continuous pipetting and incubated at ambient temperature for 1, 2, 5, 10, 20, and 30 minutes, after which the Final A260 was measured using the NanoDrop spectrophotometer.
Formamide
The formamide in liquid form of molecular biology grade (-99.5%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). 25% and 50% formamide solution with DNA were achieved by adding 10 μL and 20 μL of formamide stock solution to 30 μL and 20 μL of DNA, respectively. The mixtures were homogenized gently with pipette and incubated at room temperature, and the Final A260s were recorded with the same manner at 1, 2, 5, 10, 20, and 30 minutes.
Dimethyl Sulfoxide
The DMSO was also purchased from Sigma-Aldrich at a grade for molecular biology use (-99.9%). Three concentrations of DMSO, 25%, 50%, and 60% were employed to denature the DNA. For each concentration, 10, 20, and 24 μL of DMSO stock solution were added to 30, 20, and 16 μL of the DNA, respectively. Solutions were again homogenized gently, incubated at room temperature, and applied for the absorbance measurement at 1, 2, 5, 10, 20, and 30 minutes.
Evaluation of the Denaturation Efficiency
The denaturation capabilities of the physical or chemical treatments on DNA were investigated based on the hyperchromic effect [13], the absorbance of DNA at 260 nm will increase about 50% when the double helix structure of the DNA is denatured to two single stranded DNA. An simplified equation developed previously by Wang and Son [14], in which denaturation percentages were calculated with the numerical change of the absorbance of the DNA at 260 nm. Three variables, Initial A260, Final A260, and Blank A260 of the DNA were measured and implemented into Equation 1.
Where, Initial and Final A260 are the absorbance of the DNA at 260 nm before and after any denaturation treatments, respectively. Blank A260 is the absorbance of the free water or reagents after physical or chemical treatments. The Final A260 contained the absorbance caused by the physical methods (i.e., beads) or the reagents used in the chemical methods, it was necessary to subtract Blank A260 from the Final A260. Two μL of the DNA of each sample or the reagents were loaded onto the pedestal of the NanoDrop for absorbance measurement at 260 nm.
Renaturation of the DNA Denatured by Chemical Treatments
The denatured DNA can reformulate hydrogen bonds between complementary single strand, making it likely to reform double helix structure again. This process is called as renaturation. It may hinder the hybridization between the denatured DNA and the probe DNA. The instant concentration change of the reagents when the denatured DNA is added to the hybridization buffer may cause the unwanted renaturation. Therefore the tendency of renaturation was investigated during the hybridization process after the chemical treatments. The phosphate buffer (0.1 mol/L, pH=7.4) was used to represent the hybridization buffer. It was confirmed that the phosphate buffer had no absorbency at 260 nm and would therefore not interfere with the absorbance of the DNA at 260 nm. Post the denaturation by chemical reagents, 4 μL of the denatured DNA was added to 40 μL of phosphate buffer. The mixture was homogenized by pipetting and incubated in a dry bath incubator at 37°C to simulate the mild hybridization condition. The absorbance of the DNA at 260 nm was measured at 1, 5, 10, 15, 20, 25, 30, 60, 90, and 120 minutes during incubation for the R_Final A260 and implemented in Equation 2.
Evaluation of Renaturation
The renaturation efficiency was also calculated based on Equation 2.
Where, the R_Initial A260 is the absorbance of the DNA at 260 nm without any denaturation or renaturation treatment. The R_Final A260 is the absorbance measured after the denatured DNA being mixed with a hybridization buffer for a prolonged time. The R_Blank A260, which serves as a negative control, is the absorbance measurement of the DNase/RNase-free water treated in parallel to the DNA at 260 nm.
Results
Physical Denaturation of DNA
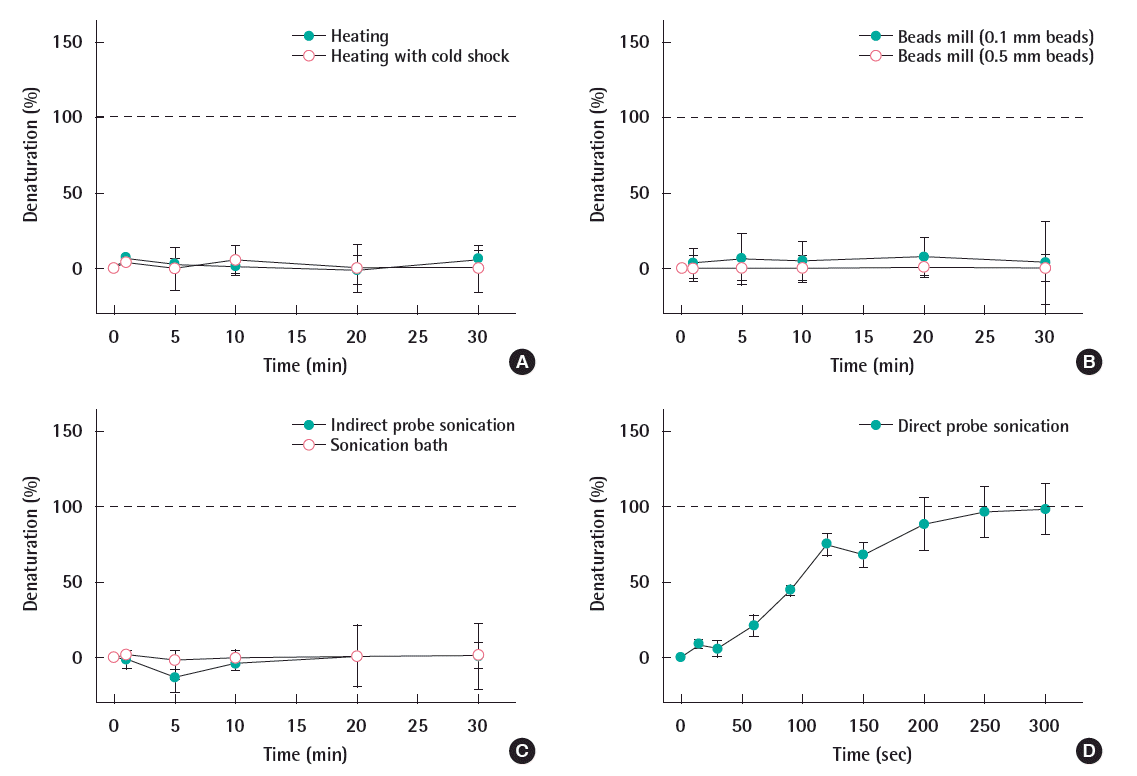
A series of physical methods were applied to the DNA fragment in order to investigate the denaturation efficiency of each method. As shown in Figure 1, the denaturation efficiency, presented as a percentage, was plotted against the time of the DNA being treated with physical methods. Figure 1A indicated the heating at 95°C, with and without the cold shock, did not denature the DNA fragment over 30 minutes. Beads mill with both 0.1 and 0.5 mm beads also did not denature the DNA for the same duration (Figure 1B). There was no denatured DNA over 30 minutes of ultrasonic bath treatment (Figure 1C open circle) and indirect sonication (Figure 1C closed circle). However, as shown in Figure 1D, the denaturation capability of the sonication increased with the time, and achieved complete denaturation capability after being sonicated for 250 seconds.
Chemical Denaturation of DNA
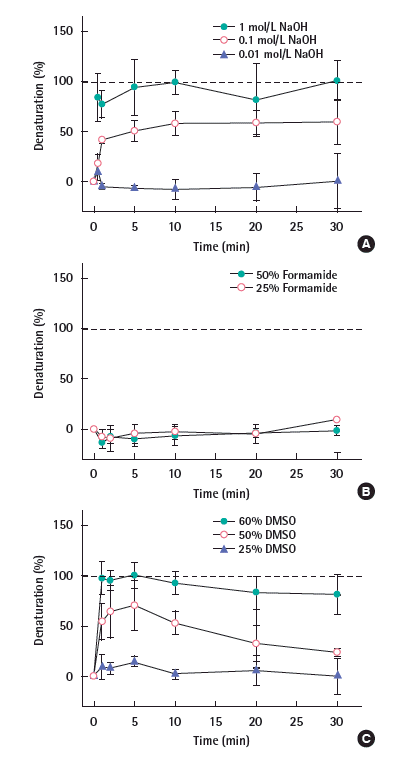
Chemical methods were also applied to denature the DNA and their denaturation efficacies are presented in Figure 2. As shown in Figure 2A, the highest concentration (1 mol/L NaOH) thoroughly denatured the DNA fragment in the early stage (i.e., 1 minute) throughout the whole process. The denaturation by 0.1 mol/L NaOH rapidly increased until 2 minutes; slowly increased until 10 minutes; and plateaued at 90% denaturation thereafter. The 0.01 mol/L NaOH denatured only around 5% of the DNA throughout the experiment. Another known chemical denaturant, formamide (25% and 50%) did not denature the DNA fragment for 30 minutes (Figure 2B). DMSO also showed the efficient denaturation result (Figure 3). 60% DMSO (Figure 2C closed circles) fully denatured rapidly within 1 minute and continued to be denatured for a 30 minutes-duration. Note that the denaturation slowly decreased to -90% over time. 50% DMSO (Figure 2C open circles) denatured 60% of the DNA was denatured within 1 minute, followed by moderately achieving 70% denaturation at 5 minutes, and then slowly decreasing up to 40% denaturation at 30 minutes. In the presence of 25% DMSO (Figure 2C closed triangles), only 10% DNA denatured throughout the whole duration.
Renaturation of the Chemically Denatured DNA
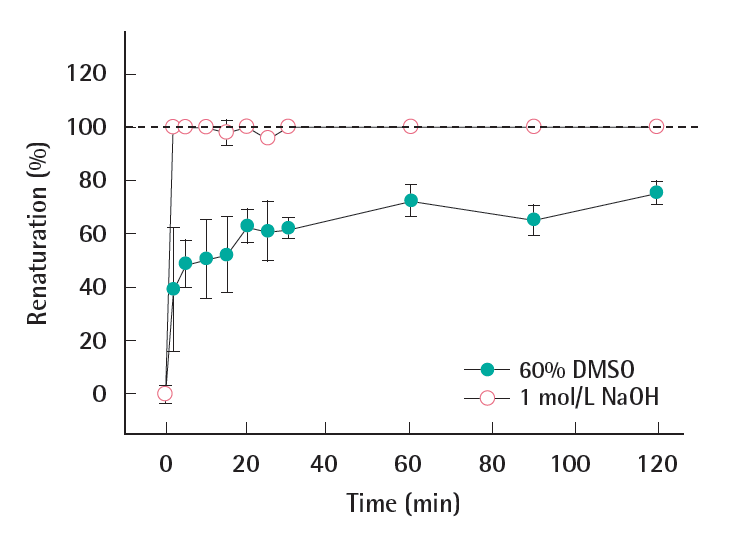
The possibility of renaturation of already denatured DNA was monitored over time under the hybridization conditions. Calculated by equation 2 and presented in Figure 3, the renaturation (%) of all denatured DNAs increased after the addition of phosphate buffer. The DNA fragment treated with 1 mol/L NaOH completely renatured as soon as the phosphate buffer was added (at 1 minute). On the other hand, the DNA denatured by 60% DMSO maintained 60% of the denatured DNA after the phosphate buffer was added (at 1 minute). Afterwards the renaturation slowly increased, however about 30% of DNA remained denatured for 2 hours.
Discussion
Heating
Theoretically the 86-bp DNA fragment will be completely denatured during the heating process at 95°C since the melting temperature (Tm) of the DNA was calculated to be 76.2°C according to Wallace et al. [15]. However, the experimental result (Figure 1A) indicated the heating methods (at 95°C), regardless of the cold shock, appeared to have no effects in the denaturation of the DNA fragment. This contradictory result can be explained by the technical limitation that the sample quantity used for the NanoDrop analysis is only 2 μL, hence the temperature of the sample probably has dropped quickly during the transfer. The sudden drop of temperature can cause an immediate renaturation effect of the already denatured DNA, making the absorption at 260 nm returned to be the same as the one before heating. Geiduschek [16] also observed the rapid DNA renaturation when the temperature decreased below the Tm. This problem can be fixed by the more sophisticated setup of heating system along with the elucidation of kinetics for this potentially rate-dependent reaction. On the other hand, a number of researchers used the heating and cooling cycle technique to manipulate the DNA in PCR [6,11,17,18]. However, unlike the PCR process that is a closed system, DNA hybridization is operated as an open system. This open system allows for the temperature of the denatured DNA to drop when it is transferred to the hybridization buffer. Therefore, heating the DNA alone may not be an adequate denaturation approach for DNA hybridization.
Beads Mill
The beads mill method is one of the well-known physical disruption techniques used to break down the microbial cell wall for the DNA extraction [19-21]. In this study, we prolonged the time of the beads mill to denature the DNA fragment and monitored the denaturation capability over time. The strong denaturation capability of the beads mill method may allow us to combine the cellular lysis and DNA denaturation; thus simplifying the sample preparation procedure for the hybridization using whole cells. However, as shown in Figure 1B, beads mill methods with 0.1 mm beads only denatured 5% of the DNA into ssDNA during the 30 minutes treatment, and the DNA was vortexed together with the 0.5 mm beads showed no denaturation. This incomplete denaturation of the DNA indicated that the disruption technique using the glass beads was not strong enough to break the double helix structure of the DNA.
Indirect Sonication
The ultrasonic bath has been used for lysing cells and breaking the chromosome DNA for lower stream experiments such as PCR or microarray [22]. In order to simplify DNA hybridization from whole cells, the denaturation capability of the ultrasonic bath was investigated. According to the absorbance of the DNA, there was no denatured DNA over 30 minutes of ultrasonic bath treatment (Figure 1C open circle). Probe sonication is a more fierce approach which is also popular in cell or DNA disruption. For the indirect probe sonication in this study, the absence of direct contact of the probe and DNA made the DNA free of possible contamination. However, the disruptive capability of the indirect probe sonication decreased significantly. As shown in Figure 1C closed circle, no DNA denaturation occurred over 30 minutes. Hence, the indirect sonication methods are not suitable for DNA denaturation.
Sonication
The direct probe sonication was carried out to denature the DNA. As shown in Figure 1D, the denaturation capability of the sonication increased with the time, and achieved complete denaturation capability after being sonicated for 250 seconds. This is likely due to the combinational effects based on heating as well as the movements of the shock waves generated during the sonication. Sonication is a commonly used fragmentation method [7,23], but it has not yet been used for denaturing DNA. Davis and Phillips [24] stabilize the DNA by controlling the temperature at 0-2°C, and observed that sonication did not denature DNA after 8 hours. In this study, the temperature increased to 82°C during the sonication. Therefore, we believe that the increase of the temperature was a crucial part during sonication treatment for DNA denaturation. Moreover, unlike the heating method, sonication may also disturb the denatured DNA and prevent the rapid renaturation. Therefore, among all the physical methods applied, the direct probe sonication was the most effective way to denature the DNA fragments.
Alkaline
The sodium hydroxide (NaOH) is a commonly used reagent to denature the DNA by increasing the pH [25-29]. At an alkaline pH, OH- groups are predominant. They remove the hydrogen- bonds-contributing protons from guanine and thymine, thus breaking the hydrogen bonds between the two oligonucleotides [27]. The result shown in Figure 2A is consistent with the previous studies, where the NaOH concentration of 0.15 [26], 0.2 [23,25] and 0.3 mol/L [29] were selectively used for DNA denaturation. Those concentrations are within the range between our second highest (0.1 mol/L) and the highest concentration (1 mol/L), which have shown 90% and 100% denaturation capability, respectively. To ensure and leverage the complete denaturation in the follow-up study for gDNA, the 1 mol/ L NaOH can be selected as an effective chemical denaturation method.
Formamide
The formamide is known for its ability to lower the Tm of DNA [30], thus the DNA denatures in the lower temperature than the melting temperature. However, neither of the two concentrations of formamide (25% and 50%) denatured the DNA fragment for a prolonged time (i.e., 30 minutes) (Figure 2B) at room temperature (i.e., 25°C). McConaughy et al. [31] found the rate of reduction for the Tm by formamide was 7.2°C per 10 percent increase of the formamide; Later, Hutton [32] reported that the formamide lowers the Tm by 6°C per 10% increase of the formamide. According to those studies, the original Tm (76.2°C) of our DNA fragment can decrease down to 30-36°C in the presence of 50% formamide [31]. This means the formamide was not able to decrease the Tm to ambient temperature (i.e., 25°C), thus failed to denature the DNA fragment at ambient temperature.
Dimethyl Sulfoxide
The DMSO is another denaturation reagent known for lowering the Tm of the DNA. As shown in Figure 2C, the denaturation capability of the DMSO became more pronounced with higher concentrations at ambient temperature. According to Markarian et al. [4], the Tm of DNA decreased from 79°C to 54°C in a 20% DMSO solution; that is a 12.5°C decrease in Tm for every 10% increase of the DMSO. Using the same interpretation, the Tm (76.2°C) of the DNA fragment in this study would decrease up to around 1°C in 60% DMSO solution. Similarly, the current Tm would decrease to 14°C, 45°C in the presence of 50%, 25% DMSO, respectively. As a result, in the presence of 60% DMSO (Figure 2C closed circles), the DNA fully denatured rapidly within 1 minute and continued to be denatured for a prolonged time (30 minutes). In the presence of 50% DMSO (Figure 2C open circles), 70% of the DNA was denatured promptly within 1 minute, followed by moderately achieving 95% denaturation at 5 minutes, and then slowly decreasing up to 40% denaturation at 30 minutes. The decrease of denaturation level over time is possibly due to the formation of H2O-DMSO hydrates [5], thus resulting in less DMSO interruption of the DNA hydrogen bonds. In the presence of 25% DMSO (Figure 2C closed triangles), only 10% DNA denatured throughout the whole duration. It is evident that the estimated Tm (45°C) for 25% DMSO is higher than the temperature at which the experiment was implemented. To guarantee the complete denaturation, 60% DMSO can be selected for further denaturation on gDNA. In summary, it is concluded that the most effective method for the DNA fragment is to be the sonication for more than 2 minutes, 1 mol/L NaOH or 60% DMSO treatment for more than 1 minute.
Particularly for the chemical treatment, the neutralization and/ or the dilution of chemicals (i.e., 1 mol/L NaOH, 60% DMSO) can be caused by additional hybridization buffer. The result shown in Figure 3 indicates the pH change is critical to the success of denaturation when the alkaline method is being used. The renaturation of the DNA is likely due to the decrease in the chemical concentration under hybridization conditions as compared to the same concentration under denaturation conditions. The results above indicated that the denaturation with 60% DMSO had a more persistent effect on the DNA denaturation as compared to the alkaline treatment, with regards to the ssDNA stability under hybridization conditions. Therefore, it was concluded that 60% DMSO was the most adequate chemical denaturation method for dsDNA fragment.
In summary, a series of physical and chemical denaturation methods were implemented on 86-bp dsDNA fragment. Among all the physical methods applied, the direct probe sonication was the most effective way to denature the DNA fragments. Examining various types and concentrations for the chemical treatment, 60% DMSO was the most adequate denaturation method and it was not fully renatured to dsDNA afterwards.