INTRODUCTION
Neurodegenerative diseases increasingly prevail in our communities recently due to several causes such as aging, heritage and heavy metals exposures. Manganese chloride (MnCl2) is one of heavy metals for causing neurogenerative dysfunction which is similar to but somewhat different from idiopathic Parkinson's disease (PD) [1].
MnCl2 is widespread in our everyday environment and plays pivotal roles in many functions. Required amount of MnCl2 can be beneficial for living organisms through regulating enzymatic synthesis and promoting hematopoiesis [2,3]. It is also a fundamental ingredient for making fungicides, steel, and welding metals and many industry products [4]. However, overexposure to MnCl2 may cause neural dysfunction in humans. The symptoms are bradykinesia, rigidity, tremor [5] which are the same sort of those in PD. The mechanism of cell death induced by MnCl2 toxicity have not been understood clearly so far though several hypothesis of Endoplasmic Reticulum (ER) stress and mitochondria disorders have been reported.
The ER stress has been known to play a role in many neurodegenerative diseases as well as other diseases. It is caused from the accumulation of excessive unfold protein response (UPR) which leads to cell apoptosis [6]. Apoptosis due to ER stress has two main pathways, transcription pathway and a caspase dependent factor.
The malfunctioning of Mitochondria is associated with a number of neurodegenerative diseases as much as ER stress. Mitochondria are often called "cellular power plants" since it generates most of the cell supply of adenosine triphosphate (ATP), an important energy source, which is involved in the control of cell cycle and cell growth [7] as well as cell signaling, cellular metabolism and cell death. Also, Mitochondria play a critical role in many metabolic systems such as regulation of membrane potential [8], apoptosisprogrammed cell death [9,10]. It can transiently store calcium for the cell homeostasis which is primarily driven by mitochondrial membrane potential [11]. Mitochondria may leak some amount of high-energy electrons in the respiratory chain to form reactive oxygen species (ROS). This can lead to mutate mitochondrial DNA after resulting in significant oxidative stress in the mitochondria.
In this paper, we investigate whether ER stress and mitochondria malfunction would be related to MnCl2 toxicity.
MATERIALS AND METHODS
I. Materials
Dulbecco's modified eagle medium (DMEM), fetal bovine serum (FBS), trypsin, and other tissue culture reagents were purchased from Life Technologies, Inc. (Gaithersburg, MD, USA) MnCl2, sucrose, 4-(2-hydroxyethyl)-1-piperazinee-thanesulfonic acid (HEPES), ethylene diamine tetra acetic acid (EDTA) and other reagents were purchased from Sigma. 3, 3-dihexyloxacarbo-cyanine iodide (DiOC6) and 2, 7-dichlorofluorescin diacetate (DCF-DA; Molecular Probes) were from molecular probes (Eugene, Oregon, USA). All of the other chemicals were purchased from either Sigma or Aldrich (St. Louis, MO, USA) and stored according to the manufacturer's instructions. All reagents were of analytical grade, and plastic wares were obtained from Falcon Inc. (Franklin, NJ, USA). GRP78, C/EBP homologous protein (CHOP), β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Phosphorylated eukaryotic initiation factor 2α(p-eIF-2α) was provided by Cell Signaling Technology Inc. (Danvers, MA, USA)
II. Cell Culture and Viability
SK-N-MC human neuroblastoma cells were obtained from the American type culture collections (Manassas, VA, USA). The SK-N-MC neuroblastoma cell lines were maintained in DMEM containing 10% (v/v) heat-inactivated FBS, penicillin G (100 U/mL), streptomycin (100 mg/mL), and L-glutamine (2 mM). Cell viability was assessed with the trypan blue exclusion assay and calculated by dividing the non-stained (viable) cell count by the total cell count (3×106).
III. Fluorescent Staining of Nuclei
For apoptosis studies, the experiments were performed after treating SK-N-MC cells with MnCl2. In the cells, nuclei were stained with chromatin dye (Hoechst 33258). Briefly, cells were fixed with 3.7% paraform aldehyde for 10 minutes at room temperature, rinsed twice 5 minutes with PBS, and incubated with 10 µM Hoechst 33258 in PBS at room temperature for 30 minutes. After three washes in PBS, the cells were observed under a fluorescence microscope (MPS 60, Leica). Morphological changes of nuclei were observed under a fluorescence microscope. To evaluate apoptotic cell death, Hoechst 33258 was used to visualize nuclear morphology and the number of cell death was counted.
IV. Determination of Caspase-3 Activity
The SK-N-MC cells (3×106) were washed with PBS and incubated for 30 minutes on ice with 100 mL of a lysis buffer (10 mM Tris-HCl, 10 mM NaH2PO4/NaHPO4, pH 7.5, 130 mM NaCl, 1% triton1 X-100, and 10 mM sodium pyrophosphate). The cell lysates were spun, the supernatants were collected and the protein concentrations were determined using the BCA method. For each reaction, 30 µg of protein was added to 1 mL of a freshly prepared protease assay buffer (20 mM HEPES pH 7.5, 10% glycerol, 2 mM dithiotreitol) containing 20 mM of AC-DEVD-AMC (N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin) (BD Biosciences Pharmingen).
The reaction mixtures in the absence of cellular extracts were used as the negative controls (fluorescence background). The reaction mixtures were incubated for 1 hour at 37℃, and the level of aminomethyl-coumarin liberated from AC-DEVD-AMC was recorded using a spectrofluorometer (Hitachi F-2500) at an excitation wavelength of 380 nm and an emission wavelength range of 400-550 nm. The data were recorded as the integral of the relative fluorescence intensity minus the background fluorescence.
V. Cytofluorometric Assessment of Mitochondrial Membrane Potential
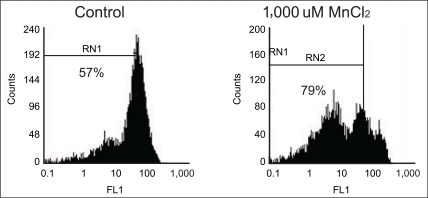
Mitochondrial membrane potential was measured as described in [12]. Briefly, a stock solution of DiOC6 (4 mM/L) was prepared in ethanol and stored in small aliquot lots at -20℃; working solution (dilution 1:2,000 for DiOC6) was diluted in an experimental medium (DMEM) immediately before use. A total of 5×105 cells were incubated in DMEM that contained 100 nM DiOC6 at 37℃, and were analyzed using a PAS cytofluorometer (Partec, Münster, Germany) equipped with Partec software. Forward and side scatters were gated for the major population of normally sized cells and a minimum of 10,000 cells were analyzed. The fluorescent probe DiOC6 was excited using a 488 nm argon laser, and emissions were collected through an FL1 detector fitted with a 525±5 nm band pass filter.
VI. DCF-DA assay
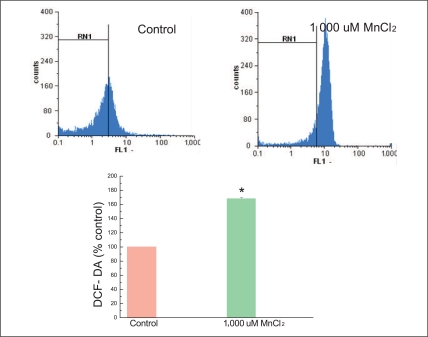
For measuring ROS, DCF-DA assay was performed as described [12]. The cells were incubated with 1 mM MnCl2 for 24 hours. After harvest, cells were treated with 100 µM DCF-DA at 37℃ for an additional 30 minutes. After chilling on ice, cells were washed with cold PBS, were removed by scraping, and then were resuspended at 1×106 cells/mL in PBS containing 10 mM EDTA. The fluorescence intensities of DCF-DA formed by the reaction between DCF-DA and the intracellular ROS of >10,000 viable cells in each sample were analyzed by PAS Vantage flow cytometry (Partec, Münster, Germany) with 488 nm excitation and 525 nm emission. Data were collected and analyzed using Becton Dickinson Partec software. The experiments were repeated at least three times, and three experiments were included in the histogram data.
VII. Western Blotting Analysis
Cell lysates (50 µg of protein) were added to an equal volume of 3×sample buffer, heated to 98℃ for 5 minutes, and then separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). After electrophoresis, the proteins were transferred onto a nitrocellulose membrane using a semi-dry electrophoretic transfer system (BIO-RAD). The membrane was incubated in 5% dry milk at room temperature for 1 hour. The membranes were then incubated with the primary antibodies at room temperature for 2 hours. The antibodies were recognized using anti-mouse or anti-rabbit IgG1 secondary antibodies linked to horseradish peroxidase at room temperature for 60 minutes. The immunoreactive bands were visualized using an enhanced chemiluminescence (ECL) kit (Amersham) and exposed to LAS 3,000 (Fujifilm, Japan).
VIII. Preparation of Homogenates
Cells were harvested with trypsin and centrifuged at 3,000 ×g for 5 minutes at 4℃. The pellet was washed 2 times with PBS and gently homogenized in buffer A (250 mM sucrose, 10 mM HEPES, 1 mM EGTA, 1 mM DTT, 2 mM MgCl2, pH 7.4) with 40 uL/mL protease inhibitor and 10 uL/mL phosphatase inhibitor using a loose-fitting dounce (Teflon-glass) homogenizer. The homogenates were rapidly placed in eppendorf tubes and then centrifuged at 1,000×g for 10 minutes at 4℃ to remove nuclei, debris, and unbroken cells. The supernatant was centrifuged at 1,000×g for 5 minutes at 4℃, and then the supernatant was centrifuged at 13,000×g for 20 minutes at 4℃ to precipitate mitochondria.
IX. Mitochondrial Complex I Assay
Mitochondrial complex I activity was measured as described in [13] using the rotenone-sensitive rate of NADH oxidation (at 340 nm and 30℃) with modification by Ragan [14]. The reaction mixture contained: 25 mM potassium phosphate pH 7.2, 0.2 mM NADH, 10 mM MgCl2, 1 mM KCN, 2.5 mg fat-free BSA, and approximately 50 µg of protein in a final volume of 1 mL. The reaction was initiated by addition of CoQ1 (50 µM final concentration) and read at 340 nm against a blank containing all the components except CoQ1. After 5 minutes, 10 µL of 0.5 mM rotenone was added to the cuvettes and the inhibition rate was measured for additional 5 minutes.
X. Mitochondrial Complex II-III Assay
Mitochondrial complex II-III activity was measured as described [13] as the antimycin-A-sensitive rate of cytochrome c reduction (at 550 nm and 30℃) using succinate as the substrate. The reaction mixture contained: 100 mM potassium phosphate pH 7.4, 0.3 mM potassium EDTA, 1 mM KCN, 100 µM cytochrome c, and approximately 50 µg of protein in a final volume of 1 mL. The reaction was initiated by addition of 20 µL of 1.0 M succinate. After the assay had run for 5-7 minutes, 10µL of antimycin- A (2 mg/mL) was added to test cuvettes and inhibited rate was measured for a further 5 minutes.
XI. Mitochondrial Complex IV Assay
Mitochondrial complex IV (EC 1.9.3.1) activity was measured as described [13] by the rate of cytochrome c oxidation (at 550 nm and 30℃) using the method of Wharton and Tzagoloff [15].
The reference cuvette contained: 10 mM potassium phosphate pH 7.0, 50 µM reduced cytochrome c, and 1 mM potassium ferricyanide in a final volume of 1 mL. The test cuvette contained: 10 mM potassium phosphate pH 7.0 and 50 uM reduced cytochrome c. The reaction was initiated by adding approximately 25 µg of protein to the test cuvette. The first-order rate constant (k) was calculated from the difference between the natural logarithms of the absorbance at t = 0 and at three time points, 1, 2, and 3 minutes after adding the protein sample. The mean of these calculated values was then taken to be k and the activity expressed in k min-1 mg protein-1.
XII. Protein Determination
Protein concentrations of the homogenates were determined using the method of Lowry [16] with bovine serum albumin (BSA) as a concentration standard.
RESULTS
I. MnCl2-Induced Apoptosis Via ER Stress
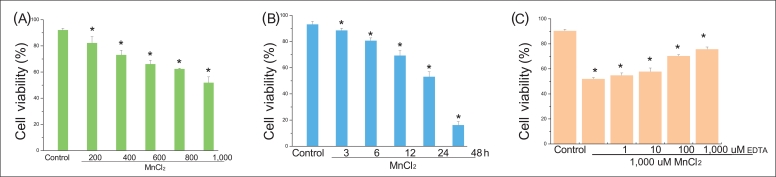
To determine the relationship between ER stress and MnCl2 toxicity, we examined cell viability including morphologic change via staining with Hoechst 33258 as wells as the expression of GRP78, CHOP, P-elf-2α. Figure 1 A shows that that cell viability with 200 uM MnCl2 is 82.26% (±4.86) but that with 1,000 uM MnCl2 is 51.91% (±4.36). The results show that higher concentration of MnCl2 decreases the cell viability. Figure 1B presents the relationship between cell viability and time, where three hours of exposure shows 88.57% (±1.60) of cell viability, however, they are 53.13% (±3.73) at 24 hours and 16.21% (±2.64) at 48 hours of MnCl2 exposure, respectively. Exposure of 48 hours by MnCl2 causes most of cells to be dead. Both increments of dose and time prompt the increments in the number of dead cells. These results lead to a conclusion that the cell death caused by MnCl2 is dependent upon both dose and time. Figure 1C shows that cell viability with 24 hours MnCl2 exposed is 51.98% (±1.05) and that of pretreatment with EDTA shows 75.75% (±1.71); treatment with EDTA increases the cell viability about 147% compared to that of MnCl2. This fact leads to a conclusion that EDTA is effective for the inhibition of the MnCl2-caused cell death.
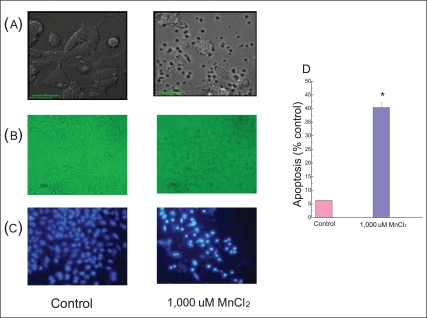
We tested with 1,000 µM MnCl2 for the next experiments to observe the apoptosis since necrosis takes place at higher concentrations of MnCl2 [17]. The cells were incubated for 24 hours. It resulted in the death of almost all of cells (Figure 1B). Figure 2 presents nuclear shrinkage, an apoptotic parameter, which shows 40.4% (±1.9) of the apoptosis compared to control cells.
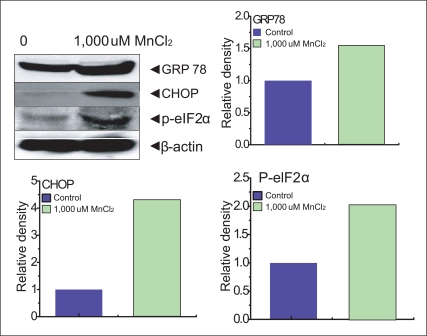
It was reported in the previous study [18] that ER stress induced by MnCl2 occurred via the activation of multiple caspases. We performed experiments on the expression of an ER stress gene, GRP78 and p-elf-2α in SK-N-MC cells. The p-elf-2α is phosporylated pancreatic ER kinase RNA like ER kinase (PERK) which attenuates the initiation of translation in response to ER stress. Our experiment showed that 154% of GRP78, 420% of CHOP, 202% p-elf-2α expression were presented compared to each control (100%) (Figure 3).
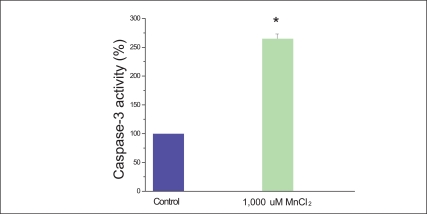
One of proapoptic proteins, CHOP which facilitates the restoration of proper protein folding within the ER, initiates cell death through the activation of caspases. The report of Morishima et al. [19] suggested that upon activation, caspase-12 translocates from the ER to the cytosol where it directly cleaves pro-caspase-9 which in turn activates the effecter caspase-3 [19]. In our experiment, caspase 3 activity protein is increased by 265±8.1% of the cell treated by 1,000 µM MnCl2 compared to the control (Figure 4). Cells treated with 1,000 µM MnCl2 clearly activated caspase-3 so that cell death was caused by apoptosis.
II. MnCl2-Induced Apoptosis Via Mitochondria Dysfunction
We tested whether cell death induced by MnCl2 was associated with mitochondria dysfunction through ROS generation, mitochondria membrane potential, mitochondria complex activities. Cells were treated with high dosage, 1,000 µM MnCl2 and incubated for 24 hours to process the above experiments. Our study showed that MnCl2 induced intracellular ROS produced 168% (±2.3%) compared to that of the control cells (100% on base line) (Figure 5) and MnCl2 induced neurotoxicity significantly dissipated 48.9% of mitochondria membrane potential compared to the control cells (Figure 6).
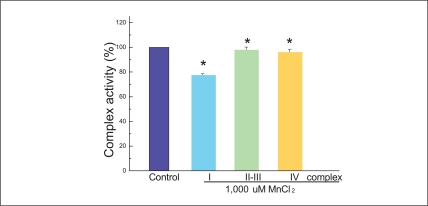
Figure 7 presents that mitochondria complex I is decreased by 22.1±0.73% of the cells treated by MnCl2. However, the cells treated by MnCl2 do not affect complex II, III or IV activities. In intrinsic activation, cytochrome c from the mitochondria forms a complex with caspase-9, apoptosis-activating factor 1 (Apaf-1), and triggers caspase-9 activation. The activation of caspase-9 and ATP processes procaspase-3 [20-22]. Then, MnCl2 toxicity causes also apoptosis induced mitochondria malfunction.
DISCUSSION
Our goal is to examine whether ER stress mediated processes may converge into intrinsic, mitochondrial pathway apoptosis in human blastoma SK-N-MC cells treated by MnCl2.
It is known that cell death is normally caused by two mechanisms: necrosis and apoptosis. Necrosis is a mode of cell death that occurs under extremely physiological conditions to damage plasma membrane and makes plasma membrane to be evoked. Necrosis starts when cell homeostasis cannot be maintained and occurs with influx of water and extracellular constituents. It eventually leads to cell swelling and rupture in intracellular mitochondria.
However, apoptosis is a process for a cell to lead to its death from normal physiological conditions. It is related with two mechanisms such as ER stress and mitochondria dysfunction.
The ER stress due to the damage of Ca2+ homeostasis [23], inhibition of protein N-linked glycosylation, expression of mutant proteins cause the accumulation of misfolded proteins in the ER lumen [24]. The UPR alleviates ER stress with several factor such as induction of ER- chaperones, initiation of a degradation system and attenuation of protein synthesis [6]. One of ER-chaperones, GRP78 activates the cells treated by MnCl2 (Figure 3). ER also is involved in stress-mediated apoptotic pathways, CHOP [25]. Figure 3 shows the expression of CHOP when the cell was treated with 1,000 µM MnCl2 for 24 hours. The cells also activate p-eIF-2α which is one of proteins related with ER stress (Figure 3). The ER stress activates caspase-12, thereby triggering an ER-stress-specific cascade for implementation of apoptosis [26]. Also, when ER stress is prolonged, it may lead to the processing of the ER-resident protease procaspase-12 as well as the activation of calpain, caspase-3, caspase-6, and apoptosis in AD [26]. Administration of 1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) which causes a severe and irreversible Parkinsonian symptoms in humans induces activation of caspases, including caspase-3, 8, 9, and 11 [27-29]. Caspase-3 is activated in the apoptotic cell both by extrinsic death ligand and intrinsic mitochondrial pathways [30,31](Figure 4).
Mitochondria are the essential generators of intracellular ROS. Excessive production of ROS, formation of PTP, release of small proteins such as cytochrome c and apoptosis-inducing factor (AIF) cause mitochondria dysfunction, leading to trigger to initiate apoptosis. Released cytochrome c binds Apaf-1 and activates the caspase cascade, caspase-9 activation. Caspase-9 leads to stimulating activation of other caspases, such as caspase -3,-6,-7, which in turn head up a series of apoptotic events, finally leading to cell death [32-34].
In our experiment, Figure 5 shows the fact that MnCl2 generates ROS spontaneously from mitochondria. MnCl2 causes the dissipation of mitochondrial membrane potential (Figure 6) significantly by activating NADPH 314 K Oxidase. Also, MnCl2 inhibits the activity of mitochondrial electron transport chain complex I (Figure 7) and decreases cellular ATP content.
Finally, MnCl2 induces caspase-3 activity, a final executioner of apoptosis followed by cell death (Figure, 2B, 2C and 4).
Our finding about the different roles of MnCl2-induced apoptosis may provide interesting insight into the mitochondrial and ER involvement in the process of different apoptotic programs. Caspase-3-dependent apoptosis of dopaminergic neurons might be also the essential catalyzing event that triggers subsequent progressive Manganism. Manganese toxicity can lead to neuronal apoptosis through the activation of intracellular apoptosis pathway. That means apoptosis might additionally contribute to the neural death in Manganism. But further studies might be necessary to uncover which other mechanisms are involved in the neural death in Manganism.
We hope this study would help further understanding of the cell death mechanisms in Manganism, particularly in regard to possible toxic effects impacted by occupational environment.