INTRODUCTION
Marijuana (Cannabis sativa) is one of the most frequently abused drugs in Korea, second only to methamphetamine [1]. The history of marijuana was originated in BC 4,000 China from hemp fibers. In oriental medicine, debarked seeds of cannabis, which do not contain any hallucinogenic compounds, have been used to relieve constipation. The hallucinative effect is usually improved when dried or its flower, stem, seed and cannabis leave are burned [2].
With the exception of Δ9-tetrahydrocannabinol (THC), the major psychoactive component, marijuana smoke is composed of qualitatively similar ingredients to those that are present in tobacco smoke (e.g., carbon monoxide, cyanide, acrolein, benzene, vinyl chlorides, aldehydes, phenols, nitrosamines, and various types of polycyclic aromatic hydrocarbons) [3,4]. In view of this similarity in the chemical contents of the smoke from both substances, concerns have been raised about possible harmful effects of marijuana on the lung. Marijuana smoke has been associated with numerous adverse pulmonary effects in humans including chronic bronchitis, edema, and mucus hypersecretion [5]. In addition, marijuana smoke has been linked to mutagenicity and genotoxicity in many studies. In human studies, researchers have noted an increase in hypoxanthine-guanine phosphoribosyltransferase gene mutations, which is used as a model gene to investigate gene mutations, in the lymphocytes of marijuana-smoking mothers and their newborns [6] and an increase in DNA strand breaks in macrophages lavaged from the lungs of marijuana smokers [7]. Marijuana smoke condensates (MSCs) have been shown to be mutagenic in Ames test, and induction of cytogenetic changes in mammalian cells following exposure to MSCs has been observed both in vivo and in vitro [8,9]. There have been several explanations for the toxic mechanisms caused by marijuana smoke. Sarafian et al. [10] reported that marijuana smoke induces the generation of reactive oxygen species (ROS) in human endothelial cells. In addition, THC was found to induce oxidative stress in many cell lines via the central cannabinoid (CB1) receptor pathway [11]. In the present study, we evaluated a possible role for p53 in lung damage caused by MSCs, including genotoxic effects and apoptosis. The p53 tumor suppressor is a transcription factor that regulates gene expression relating to the cell cycle, DNA repair and apoptosis. In particular, the activation of p53-regulated genes is responsible for determining whether or not damaged DNA moves forward to DNA repair, or is disposed of via cellular apoptosis. However, continuous apoptosis can result in prolonged inflammation and delayed systemic repair because toxic contents in apoptotic cells may be released [12,13]. To meet our goal, two cell lines were chosen: p53-wildtype (WT) H460 and p53-null H1299 human adenocarcinoma cells. H460 and H1299 cells have been used as models p53 positive and negative cell lines [14,15]. We determined genotoxicity, oxidative stress and apoptosis in both cell types after exposure to MSCs.
MATERIALS AND METHODS
I. Preparation of MSCs
Non-filtered marijuana cigarettes were prepared from dried marijuana plants (National Forensic Service, Korea) using a manual cigarette-stuffing machine (Privileg, Germany). Twenty-three marijuana cigarettes (average weight 646 mg) were burned and MSCs were collected on a fiber filter (10 cm×10 cm, Sibata, Tokyo, Japan) in a smoking chamber using a high-flow vacuum pump connected to a cascade impactor (Dusturbo, Seoul, Korea) at a flow rate of 28.3 L/min. The dry weight of the MSCs was measured and the 23 non-filtered marijuana cigarettes generated 339 mg of total MSCs. Pieces (1 cm×1 cm) were cut from the filters and each was immersed in a volumetric flask. The MSCs were extracted by sonication with methylene chloride, filtered, and evaporated to dryness. The residue was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 100 mg/mL and stored at -20℃ prior to cell exposure.
II. Cell Culture
p53-WT H460 cells, with a p53-WT gene, and p53-null H1299 cells, lacking the p53 gene, were obtained from the American Type Culture Collection (ATCC; HTB-177™ and CRL-5803™, respectively). These cells were grown in RPMI1640 supplemented with 5% fetal bovine serum (FBS), penicillin (100 units/mL) and streptomycin (100 µg/mL) at 37℃ in an atmosphere of 5% CO2 / 95% air under saturating humidity.
III. In Vitro Single Cell GEL Electrophoresis Assay
To evaluate the genotoxicity of the MSCs, we performed the in vitro single cell gel electrophoresis assay (comet assay) as described by Singh et al. [16]. H460 and H1299 cells (20×104 cells/well) were plated on 6-well plates and the culture was maintained at 37℃ in a 5% CO2. After 24 hours incubation, the cells were exposed to MSCs for 24 hours. The treated cells were then trypsinized to produce a single-cell suspension, washed in ice-cold phosphate-buffered saline (PBS, pH 7.4) and resuspended in 0.7% low melting point agar (Quantum Biotechnologies Inc., Quebec, Canada) in PBS at a concentration of 5×104 cells/mL. A 150 µL aliquot of this suspension was pipetted onto precoated glass slides, covered with a glass coverslip, and then the agar was allowed to set at 4℃. The sample was placed in lysis solution. For the alkaline comet assay, cells were lysed in pH 10 lysis solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris-HCl, 1% Triton X-100) at 4℃ for 60 minutes. And the slide was covered with 0.7% low melting point agar and the agar allowed to set at 4℃. The lysed cells were washed and allowed an unwinding time of 30 minutes in electrophoresis buffer (1 mM Na2EDTA, 300 mM NaOH pH 13). Then electrophoresis was carried out for 30 minutes at 25 V on ice and the cells were neutralized with 0.4 M Tris-HCl (pH 7.5) for 10 minutes. The cells were stained with ethidium bromide (2 µg/mL) just prior to the image analysis. DNA migration was evaluated by fluorescence microscopy (excitation filter 515 to 560 nm and a barrier filter of 590 nm; Leica DMLB, Leica, Wetzlar, Germany) in conjunction with a digital camera. The images were assessed by an auto image analysis system (Komet version 5.0, Kinetic Imaging Ltd., Liverpool, UK). The olive tail moment (tail distance×percentage of DNA in tail) was used to quantify the DNA damage, based on the random scoring of 100 nuclei per slide.
IV. In Vitro Cytokinesis-block Micronucleus Assay
To evaluate the macro-genotoxic effect of MSCs, we performed the in vitro cytokinesis-block micronucleus (CBMN) assay, which offers a good alternative to the conventional chromosome aberration technique [17]. In the micronucleus (MN) assay, p53-WT H460 and p53-null H1299 cells were seeded into 8-well-chamber slides at a density of 1.8×104 cells/well and cultured for 24 hours until they reached 70%-80% confluence. The treatment medium was prepared by adding MSCs to medium. For each independent test, at least two slides were prepared. To restrict the analysis to cells that had passed through their first division, the cells were incubated in growth medium containing 3 µg/mL cytochalasin B [18]. One cycle after cytochalasin B was added, the growth medium was removed from the 8-well-chamber slide by aspiration. The slide was washed twice with the same amount of PBS and left for 5 minutes at 4℃ after 1% sodium citrate was added. The slide was then placed in fresh fixative (99:1=methanol:acetic acid) for 20 minutes at room temperature, and subsequently left on a clean bench to air-dry before being placed in ribonuclease A (10 µg/mL in 2×saline-sodium citrate (SSC)) for 5 minutes at 30℃. The slide was then rinsed in 2×SSC (0.03 M sodium citrate, 0.3 M NaCl) and left on a clean bench to air-dry. After thorough air drying, the slide was stained with 5% Giemsa solution for 14 hours. According to the MN scoring criterion, a score of 1,000 binucleate cells per independent culture was obtained. Positive mutagenic effects of the samples were defined as a threefold or higher increase in the MN frequency over the control with at least one of the tested doses [19].
V. Determination of ROS Generation
The methods of Hussain et al. [20] and Fotakis et al. [21] were adopted to determine the increase in intracellular ROS in MSCs-treated p53-WT H460 and p53-null H1299 cells. Cells were seeded in 48-well plates at an initial concentration of 4×104 cells/well, and incubated for 24 hours. Cells were grown to 80% confluence on 48-well plates and exposed to MSCs for 6 hours. 2', 7'-dichlorohydrofluorescein diacetate (DCF-DA) was added for 1 hour as an intracellular ROS indicator. This assay requires the diacetate to be cleaved chemically [22]. So each well was washed twice with PBS, and lysed using 0.1N NaOH. DCF (green fluorescence) production was measured in an LS50B fluorometer (Perkin-Elmer, Shelton, CT, USA) with an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
VI. Apoptosis
The apoptosis in cells exposed to MSCs was analyzed using the Cell Death Detection ELISAPLUS (Roche Diagnostic, Montclair, NJ, USA) protocol. Approximately 104 cells/well were seeded in a 96-well plate. After 24 hours of incubation, the medium was changed to MSCs-containing medium for 24 hours. Referring to the manufacturer's instructions, at the end of the incubation period, the cells were lysed and the levels of apoptosis were determined using the Cell Death Detection ELISA assay, which measures cytoplasmic histone-associated DNA fragments (mono- and oligo-nucleosomes).
VII. p53-related Gene Expression
BEAS-2B cells (60×104 cells/well) were seeded on 6-well plates and the cultures were incubated for 6 hours at 37℃. After MSCs exposure, total RNA was isolated from BEAS-2B cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Messenger RNA species can be reverse transcribed into cDNA through the enzymatic activity of many available reverse transcriptases (RT) in the presence of dNTPs and an oligo dT primer. One microgram of mRNA isolated from cells was placed into a reaction tube with 0.5 µg/mL of random primers, 1 µL of AMV reverse transcriptase, 4 µL of 5×RT buffer, 0.5 µL of RNase inhibitor, and 1 µL of 10 mM dNTPs. The RT conditions were as follows: room temperature for 10 minutes, 42℃ for 50 minutes, and 94℃ for 5 minutes. The reaction was performed according to the manufacturer's instructions. cDNA from each of the samples was then used. Specific primers were designed and are shown in Table 1. Ribosomal RNA (18s) levels were used to normalize the results. The real-time fluorescence signal produced by the nonspecific double-stranded DNA binding dye was analyzed using Smart Cycler software. Polymerase chain reaction (PCR) was conducted using 0.1 µM of the sense and antisense oligonucleotide primers, 12.5 µL of 2×SYBR Premix Ex Taq (Takara Bio Inc., Shiga, Japan), and 1 µL of template DNA in a final volume of 25 µL. The PCR amplification was performed using a Smart Cycler (Cepheid, Sunnyvale, CA, USA). The fluorescence signals were read at the end of each extension step. The threshold cycle was determined for each sample using the exponential growth phase, and the baseline signal from the fluorescence versus the cycle number plots. To ensure that a single product was amplified, melt curve analysis was performed on the PCR products whenever each PCR run was done.
VIII. Data Analysis
SigmaPlot 10.0 (Jandel Science Software, San Rafael, CA, USA) and Excel 2007 (Microsoft, Redmond, WA, USA) were used to analyze the data. The data from each assay were expressed as mean±standard deviation. Statistical analysis was performed using the SPSS version 17.0 (SPSS Inc., Chicago, IL, USA) program. Differences between groups were tested by t-test following one-way analysis of variance (ANOVA). Statistical significance was accepted at p < 0.05 and 0.01.
RESULTS
I. Genotoxic Effects
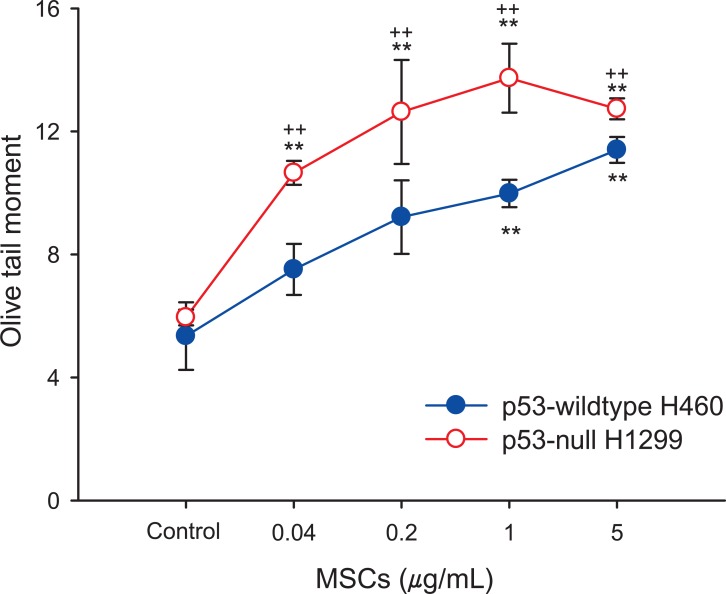
In order to investigate the genotoxic effects of MSCs, we conducted the comet assay and CBMN assay. To assess the DNA damage induced by MSCs exposure, comet assay was performed on p53-WT H460 and p53-null H1299 cells. After treatment with MSCs for 24 hours, DNA damage in the two cell lines were significantly increased in a dose-dependent manner (0.04 to 5 µg/mL), (Figure 1). At all doses employed (0.04, 0.2, 1 and 5 µg/mL), the DNA damage induced by MSCs in p53-null H1299 was statistically higher than in p53-WT H460 cells. In the CBMN assay, significant MN formation was observed in the two cell lines treated with MSCs in a dose-dependent manner (0.04 to 5 µg/mL) (Figure 2). MN formation at the highest dose (5 µg/mL) in H460 and H1299 was increased by 2.24-fold and 3.28-fold compared with those in each control group, respectively, showing the greater sensitivity of p53-null H1299 (p < 0.01).
II. ROS Formation
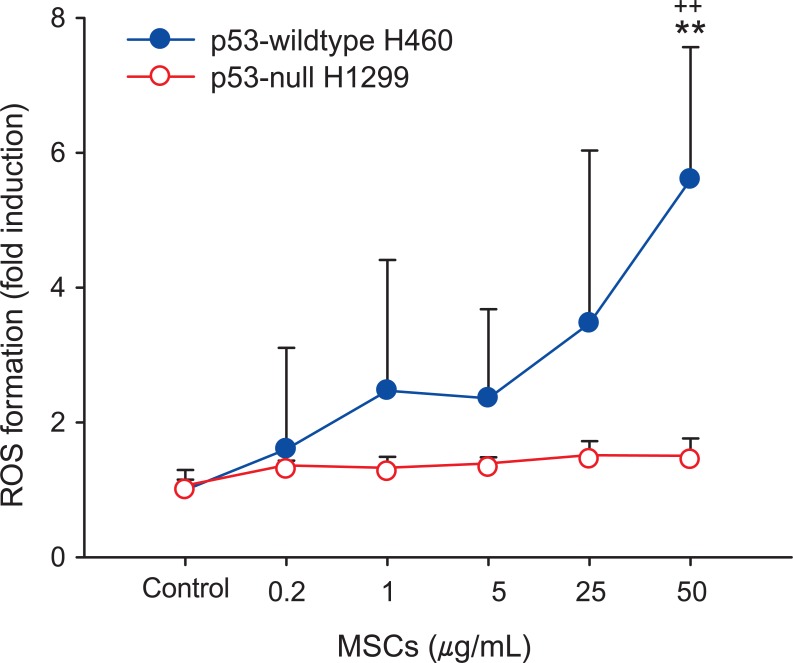
To identify p53-dependent oxidative stress by MSCs, ROS was measured in p53-WT H460 and p53-null H1299 cells. As shown in Figure 3, MSCs exposure enhanced DCF fluorescence in p53-WT H460 cells dose-dependently, whereas no significant enhancement was noticed in p53-null H1299 cells. ROS generation in p53-WT H460 cells treated with the highest concentration of MSCs was 5-fold higher in comparison with the control (p < 0.01) (Figure 3). This result confirmed that MSCs exposure enhanced ROS generation in p53-WT H460 cells.
III. MSCs-induced Apoptosis
We investigated cell apoptosis in response to MSCs exposure. A cell death detection ELISA assay was performed on p53-WT H460 and p53-null H1299 cell lines exposed to MSCs for 24 hours. The result is presented as an enrichment factor as compared to p53-WT H460 control cells (Figure 4). In p53-WT H460 cells, apoptosis triggered by MSCs showed a significant increase compared to control, but in p53-null H1299 cells, there was no apparent apoptosis induced by MSCs.
IV. p53- and Apoptosis-related Gene Expression
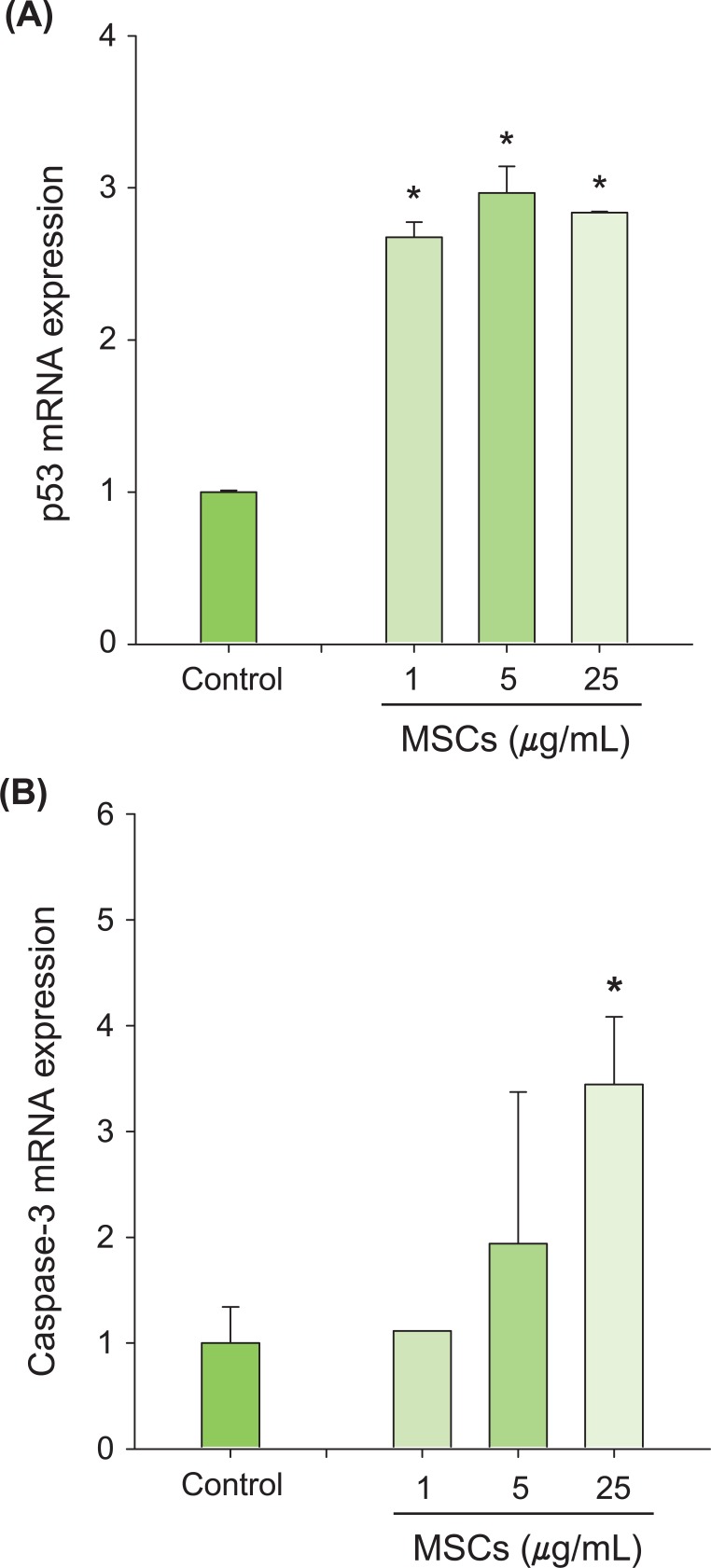
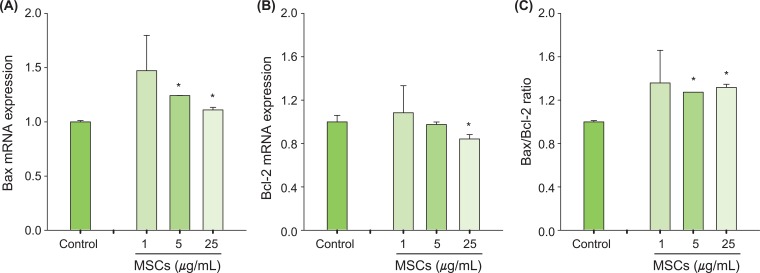
p53 and Bax mRNA levels were quantified using a highly specific, sensitive and rapid SYBR green quantitative real-time PCR (Figure 5). Treatment with MSCs for 6 hours significantly increased mRNA expression of apoptosis-related genes, including p53 (Figure 5A), and caspase-3 (Figure 5B). Expression of the apoptotic Bax gene was increased significantly (Figure 6A) and expression of the anti-apoptotic Bcl-2 gene was decreased significantly compared to control (Figure 6B). In addition, the ratio of Bax to Bcl-2 was significantly increased after MSCs exposure (Figure 6C). Collectively, these results indicated that p53 and other apoptosis-related genes are involved in MSCs-induced apoptosis.
DISCUSSION
Mutated p53 gene is considered as the most frequently involved factor in lung cancer. Simultaneous genetic malfunction in bronchial epithelial cell brings forth cytomorphological changes that proceeded to the lung cancer [23]. The p53 gene mutated in human cancers [24] is involved in control of DNA damage repair [25], apoptosis [26], and cell cycle progression [27]. The role of p53 is anticarcinogenesis by disturbing actions like apoptosis, arresting cell cycles in DNA damaged cells. Studies of particulate matters on DNA damage have focused on organic extracts as the major fraction [28]. Hundreds of components are consisting organic fraction, including some genotoxic groups of compounds such as polycyclic aromatic hydrocarbon, nitrated or oxygenated derivatives [29,30]. Therefore, to identify lung carcinogenesis induced by MSCs, we determined the genotoxic effects and apoptosis of MSCs dependent on p53 only in the organic fractions.
MSCs-induced DNA damage was dose-dependently increased in p53-WT H460 cells and p53-null H1299 cells (Figure 1). Some researchers have reported that marijuana smoke induces DNA damage by formation of DNA adducts [31]. In the CBMN assay with p53-WT H460 and p53-null H1299 cells (Figure 2), MSCs also stimulated a significant increase of MN formation in a dose-dependent manner. However, exposure of p53-null H1299 cells to MSCs produced more DNA breakage and MN formation than p53-WT H460 cells exposed to MSCs. Pfeifer et al. [25] reported that p53 is involved in control of DNA damage repair. Therefore, we suggest that the significant damage to DNA and chromosomes in p53-null H1299 cells exposed to MSCs may be due to an insufficiency in the DNA repair pathway caused by p53 deficiency.
ROS production plays an important role in apoptotic responses through p53 activation [32-34]. MSCs induced a significant increase of ROS production in p53-WT H460 cells, but not in p53-null H1299 cells (Figure 3). The difference in ROS production in the two cell lines may also be related to altered apoptosis following exposure to MSCs. p53-WT H460 cells exposed to MSCs showed higher apoptosis than p53-null H1299 cells (Figure 4). We suggest that the lack of p53 gene in p53-null H1299 cells may induce efficiencies in the apoptosis pathway dependent on p53.
To identify gene regulation related to p53-dependent apoptosis, we measured the expression of p53-related genes in p53-WT H460 cells using real-time PCR. The mRNA expression of caspase-3 and p53 was significantly increased by MSCs (Figure 5). Caspase-3 exists as an inactive enzyme and is activated early in the apoptotic process after apoptotic signaling events have occurred [35]. The stabilization and accumulation of p53 can induce up-regulation of the apoptotic Bax gene, which can induce apoptosis [36]. In addition, Bax binds to the anti-apoptotic Bcl-2 gene, which regulates the apoptotic process [37]. The Bcl-2 gene plays an important role in preventing apoptosis induced by diverse stimuli and conditions [38,39]. In this study, levels of Bax mRNA were significantly increased while that of Bcl-2 was significantly decreased by MSCs exposure (Figure 6). The ratio of Bax/Bcl-2 is important for determining a cell's susceptibility to apoptosis and up-regulation of cell death genes [40]. The ratio of Bax to Bcl-2 in H460 cells exposed to MSCs was significantly increased compared to that of the control (Figure 6C). Therefore, we suggest that apoptosis-related genes (Bax and Bcl-2) could be involved in the MSCs-induced apoptotic pathway.
In conclusion, MSCs significantly increased DNA breakages and chromosomal changes in p53-WT H460 and p53-null H1299 cells. In particular, genotoxicity was shown to be significantly higher in H460 than in H1299 cells even though the reverse was true for ROS induction by MSCs. We expect apoptosis is stimulated by Bax/Bcl-2 gene regulation in H460 cells for this reason. These results suggest that MSCs induce DNA/chromosomal damages in human lung cancer cells, and p53 plays an important role in the cellular response of MSCs.