INTRODUCTION
Breast cancer is the major cancer affecting females around the world today. In Korea, breast cancer incidence has been continuously increasing and in 2002, breast cancer became the most common form of cancers among Korean women. According to the American Cancer Society 2005 report [1], breast cancer is the most frequent cancer in women of the United States. In the United States alone, currently around 40,000 female and 1,500 male breast cancer deaths occur per annum. Current therapeutic approaches for human breast cancer include hormonal therapy with anti-estrogenic compounds, as well as surgery, radiotherapy, hyperthermia, and chemotherapy [2]. However, conventional strategies for treatment of breast cancer are still unsatisfactory and limited. Therefore, there is an urgent need to develop more effective therapeutic approaches for prevention and treatment of breast cancer.
Recently, histone deacetylase has been discovered as a potential target for the development of new therapeutic agents. Recent data suggested that histone deacethlase (HDAC) inhibitors displayed potent anti-tumor activity via cell cycle arrest, differentiation, and induction of apoptosis in the various tumor cells, whereas they showed low or negligible effects on the untransformed cells [3-8]. These selective inhibitory effects on the transformed cells compared with normal cells are not completely understood. However, they do not appear to be caused by a difference in the ability to inhibit HDAC activity because they cause a similar accumulation of acetylated histone proteins in both normal and tumor cells [9-13]. Another important characteristic of HDAC inhibitors is their selectivity in altering gene expression in transformed cells. Several studies have now revealed that fewer than 10% of expressed genes in a given malignant cell are affected by HDAC inhibitors, with a near equal number of transcriptionally active genes being suppressed as being stimulated.
The report showed that tumor-associated proteins mediated proliferation and cell cycle progression, survival factors, growth factor receptors, kinases, signal transduction intermediates, DNA synthesis/repair enzymes, shuttling proteins, transcription factors, and protease. Recently, HDAC inhibitors have become an exciting new class of drugs that have been targeted as anti-cancer agents [14-16]. A number of clinical trials have shown the utility of HDAC inhibitors in treatment of tumors. However, the accurate molecular basis for their anti-cancer activity remains largely unknown.
In this study, we tried to evaluate the anti-tumor effects of various HDAC inhibitors on MCF-7 human breast cancer. Moreover, the underlying chemotherapeutic mechanisms of them were also explored. To examine the anti-tumor effect of HDAC inhibitors, we examined the effect of HDAC inhibitors on the cell proliferation, cell cycle distribution, and apoptosis in MCF-7 human breast cancer cells. To find out the mechanism of anti-tumor activity of HDAC inhibitors, we examined the effect of IN-2001 on the expression of cell cycle regulatory protein and apoptosis-related proteins.
MATERIALS AND METHODS
I. Chemicals
HDAC inhibitors, such as Trichostatin A, IN2001, suberoylanilide hydroxamic acid (SAHA), and ((E)-N-hydroxy-3-[4-[[2-hydroxyethyl-[2-(1H-indol-3-yl0ethyl)amino]methyl]phenyl]prop-2-enamide (LAQ) were generously provided from Dr. Kim (Ewha Womans University, Seoul, Korea). Helminthosporium carbonum (HC) toxin was obtained from Sigma Chemical Co. (St. Louis, MO, USA).
II. Cell Lines and Cell Culture Conditions
MCF-7 cells were obtained from Korean Cell Line Bank (Seoul, Korea). MCF-7 cells were maintained in Minimum Essential Medium (Garlsbat, CA USA), supplemented with fetal bovine serum, insulin and penicillin-streptomycin. Cells were routinely maintained at 37℃ and in 5 % CO2.
III. Cell Proliferation Assay
Cells were plated in 96 well plates at a density of 104 cells per well. The following day, the cells were treated with chemicals. The number of cells was measured based on the modified sulforhodamine B (SRB) assay of Soto et al. [17]. Cells were treated with cold 10% trichloroacetic acid (TCA) and incubated at 4℃ for 30 minutes, then washed five times with tap water and left to dry. TCA-fixed cells were stained for 30 minutes with 0.4 % (w/v) SRB dissolved in 1 % acetic acid. Wells were washed with tap water and air-dried. Bound dye was solubilized with 10 mM tris base (pH 10.5) in a shaker for 30 minutes, Finally, optical density was read using ELISA reader (Bio-Rad, Hercules, CA, USA) at 570 nm.
IV. Fluorescent Activated Cell Sorter (FACS) Analysis
Cells were plated in 60 mm2 dishes and exposed to chemicals. Treated cells were detached using trypsin-EDTA and fixed with 70% ethanol. After centrifugation, the cells were treated with RNase A (10 µg/mL) for 20 minutes at 37℃ and stained with propidium iodide (2 µg/mL) for 30 minutes at 37℃ in the dark. The DNA content per cell was evaluated in a FACSCalibur (Becton Dickinson, San Diego, CA, USA).
V. RT-PCR Analysis
Cells were plated in 60 mm2 dishes and exposed to chemicals for 24 hours. Total RNA was extracted using trizol reagent (Invitrogen Co., Carlsbad, CA, USA). Reverse transcription was carried out on 3 µg of total RNA diluted in a 22.5 µL mixture containing 1 µL random primer (0.5 µg/µL), 1 µL dNTP (1 mM), 2 µL DTT, 4 µL RT buffer (5X), 1 µL M-MLV reverse transcriptase (200 U/µL), 0.5 µL RNasin (40 U/µL), and H2O. After incubation at 37℃ for 1 hour, the reverse transcriptase was inactivated for 10 minutes at 95℃ and cDNA was stored at -20℃ or immediately used for PCR. One µL of the synthesized cDNA was subjected to PCR amplification with primer (GAPDH; 5' ACATCgCTCA gACACCATgg3'; 5' gTAgTTgAggTCAATgAAggg 3: p21; 5' gAACTTCgACT TTgTCACCgAg3'; 5' CgTTTTC gACCCTgAgAgTCTC 3': Cyclin D1; 5' AgCCATggAACACCAgCTC3'; 5' gCAC CTCCAgCATCCAggT3' in a 10 µL reaction containing dNTP and Taq polymerase. DNA was denatured at 95℃ for 5 minutes and cycled immediately 25 times at 95℃ for 30 seconds with specific annealing temperature chosen by preliminary experiments and extended at 72℃ for 1 minute. The PCR reaction ended with 5 min incubation at 72℃. PCR products were analyzed on 2% agarose gels and the image of the band on agarose gel was quantified using CoreImager MC2000 (Sam Learmdro, CA, USA).
RESULTS
I. IN-2001 Inhibits Human Breast Cancer MCF-7 Cell Growth
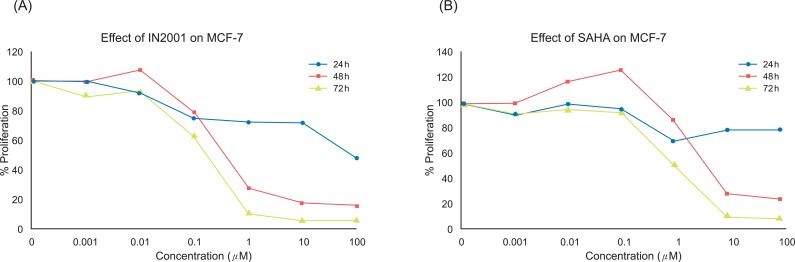
In recent years, an increasing number of structurally diverse HDAC inhibitors have been identified as an exciting new class of potential anti-cancer agents. In this study, we evaluated the anti-tumor effects of various kinds of HDAC inhibitors in the human breast cancer cells in an attempt to find out better therapeutic agents for breast cancer treatment. To determine the anti-proliferative effect of IN-2001 (Figure 1) on the human breast cancer MCF-7 cells, MCF-7 cells were treated with vehicle (0.1% DMSO) or various concentrations (0.001 µM to 100 µM) of IN-2001 for various times and then the number of cells was determined based on the SRB assay. As shown in Figure 2, IN-2001 showed an anti-proliferative effect in a dose- and time-dependent manner. The IC50 value of IN-2001 was 0.287 µM at 72 hours treatment of IN-2001 in MCF-7 cells (Table 1). MCF-7 cells showed significant growth inhibition when cells were exposed to IN-2001 for more than 24 hours, and 24 hours treatment resulted in 15-20% with up to a 10 µM IN-2001 reduction of cell proliferation compared to control. IN-2001 treatment for 48 hours and 72 hours resulted in a potent dose-dependent inhibition of MCF-7 cell proliferation (Figure 2A).
II. SAHA Inhibits Human Breast Cancer MCF-7 Cell Growth
In the next experiment, we examined the effect of SAHA as a reference HDAC inhibiting drug on MCF-7 cell proliferation and the result is shown in Figure 3. SAHA decreased the proliferation of human breast cancer cells in a time- and dose-dependent manner. MCF-7 cells showed significant growth inhibition when cells were exposed to SAHA for more than 24 hours. In MCF-7 cells, SAHA treatment for 24 hours resulted in a 15-20% reduction of cell proliferation compared to the control. SAHA treatment for 48 hours and 72 hours resulted in potent dose-dependent inhibition of MCF-7 cell proliferation (Figure 2B). The IC50 value of SAHA was 0.761 µM at 72 hours treatment of IN-2001 in MCF-7 cells (Table 1).
III. IN-2001-Induced Cell Cycle Arrest
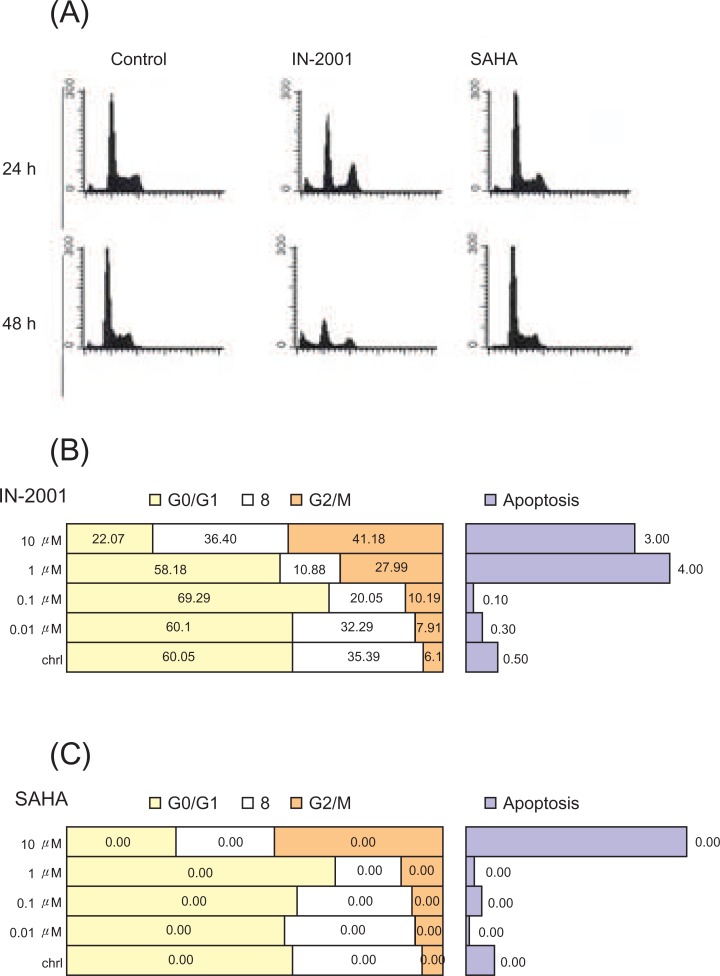
To investigate the growth inhibitory effects of IN-2001 on cell cycle alteration, we analyzed the cell cycle distribution in human breast cancer cells. MCF-7 cells were treated without or with 1 µM IN-2001 or SAHA for 24 or 48 hours, and then cell cycle distribution was analyzed by flow cytometric analysis after propidium iodide staining of cellular DNA. Representative histograms and quantitative analysis data are shown in Figure 3.
In MCF-7 cells, HDAC inhibitors except for SAHA induced G2/M arrest after 24 hours and 48 hours treatments (Figure 3A). When cells were treated with IN-2001 and SAHA for 24 hours, MCF-7 cells yielded 30.6% and 7.9% of cells in G2/M phase, respectively, whereas untreated control cells showed 6.1% of cells in G2/M phase. At 48 hours treatment, IN-2001 and SAHA accumulated 20.4% and 37.0% of cells in G2/M phase, respectively, whereas untreated control cells showed 3.9% of cells in G2/M phase. SAHA showed a different cell cycle alteration: G0/G1 arrest. At 24 hours and 48 hours treatment, SAHA accumulated 61.2% and 71.4% of cells in G0/G1 phase, respectively, whereas untreated control cells showed 53.2% and 63.8% of cells in G0/G1 phase, respectively (Figure 3B and 3C).
IV. IN-2001-Induced Apoptosis
In addition to cell cycle arrest, HDAC inhibitors have been shown to induce apoptosis in various human cancer cell lines (18 ). After 72 hours treatment of IN-2001, the extensive floating dead cells were observed in a dose-dependent manner in MCF-7 (Figure 4). These observations prompted us to examine whether the effects of IN-2001 on cell death were attributable to the induction of apoptosis. We performed FACS analysis of propidium iodide-stained nuclei to assess the presence of a hypodiploid or sub-G1 population resulting from DNA fragmentation. MCF-7 cells were treated without or with 1 µM IN-2001 for 24 or 48 hours and then stained with PI, and subjected to FACS analysis.
As shown in Figure 3, increasing exposure times of IN-2001, and SAHA led to an incremental accumulation of the apoptotic sub-G1 population in both sets of MCF-7 cells.
FACS analysis revealed that untreated MCF-7 cells showed 1.0% of sub-G1 population, but exposure of 1 µM IN-2001 for 24 hours increased the sub-G1 population to be 4.4%. When MCF-7 cells were treated with IN-2001 for 48 hours, 16.8-32.3% of apoptotic cells were observed, whereas untreated cells showed 0.5% of sub-G1 population. However, 1 µM SAHA did not show apparent sub-G1 population. As shown in Figure 4, IN-2001 and other HDAC inhibitors were found to induce cell cycle arrest and apoptosis in human breast cancer cells. Because acetylation and deacetylation of histones have been shown to play an important role in the regulation of gene expression in eukaryotic cells, cell cycle arrest and apoptosis by HDAC inhibitors might be attributable to alteration of genes involved in cell cycle progression and apoptosis. To test this possibility, we examined the effects of IN-2001 and other HDAC inhibitors on cell cycle and apoptosis-related gene expression in human breast cancer cell lines.
V. IN-2001-Induced cyclin-dependent kinase (CDK) Inhibitor, p21WAF1 Expression
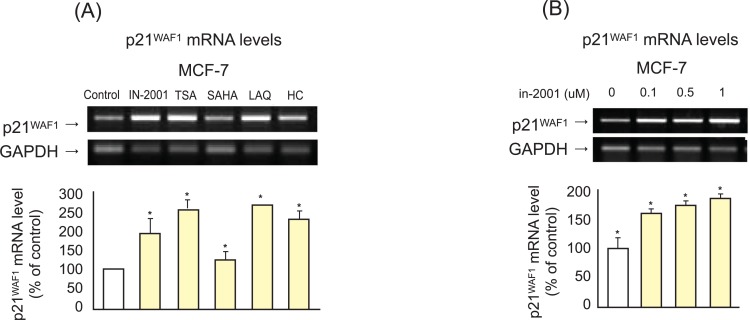
p21WAF1 is a CDK inhibitor that binds to CDK complexes and decreases kinase activity and may act as a key regulator of the cell cycle and apoptosis. Therefore, we examined the effects of IN-2001 and SAHA on the expression of p21WAF1 in human breast cancer cells. MCF-7 cells were treated with 1 µM IN-2001 or SAHA for 24 hours, and then analyzed for p21WAF1 mRNA level by RT-PCR. As shown in Figure 5A, exposure of MCF-7 cells to 1 µM HDAC inhibitors induced upregulation of p21WAF1 mRNA levels, as determined by RT-PCR analysis. In MCF-7 cells, IN-2001, TSA, SAHA, LAQ, and HC-toxin increased p21WAF1 mRNA levels by 193.6%, 255.7%, 128.6%, 266.0%, and 231.1%, respectively. A dose-dependent experiment for mRNA levels of p21WAF in MCF-7 cells treated with IN-2001 is shown in Figure 5B. In MCF-7 cells, p21WAF1 mRNA levels were induced by 152.7%, 168.8%, and 180.8% at 0.1 µM, 0.5 µM, and 1 µM IN-2001, respectively. These results suggest that upregulation of CDK inhibitors by IN-2001 may lead to cell cycle arrest, ultimately resulting in growth inhibition.
VI. IN-2001 Downregulated Cyclin D1 Expression
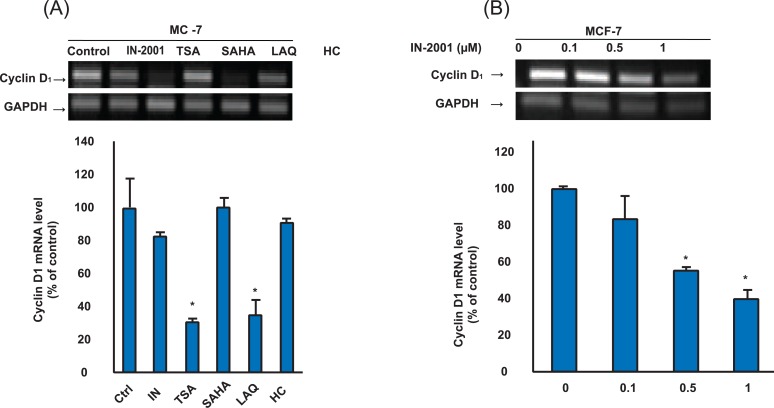
Cyclin D1 is involved in regulation of transition from G1 to S during the cell cycle. Given the critical role of cyclin D1 in cell cycle regulation, their abnormal or untimely expression could disrupt the cell cycle and therefore render them growth-promoting genes or oncogenes. Thus, cyclin D1 is considered to be a putative proto-oncogene and is overexpressed in a number of tumor types including lymphomas, breast cancer, and thyroid carcinoma. Herein, we investigated if IN-2001 and other HDAC inhibitors can regulate the expression of cyclin D1 in human breast cancer cells. MCF-7 cells were treated without or with one µM HDAC inhibitors for 24 hours, and then analyzed for cyclin D1 mRNA levels by RT-PCR analysis. As shown in Figure 6A, cyclin D1 mRNA was detectable in untreated MCF-7 and µM HDAC inhibitor treatment suppressed the expression of cyclin D1 mRNA in MCF-7 cells. IN-2001, TSA, LAQ, and HC-toxin resulted in reductions to 82.1%, 30.4%, 34.6%, and 91.1% in cyclin D1 expression, respectively. 1 µM of SAHA, however, did not change cyclin D1 expression. A dose-dependent experiment for levels of cyclin D1 in MCF-7 cells treated with IN-2001 is shown in Figure 6B. The expression of cyclin D1 mRNA was downregulated in a dose-dependent manner by treatment with doses of 0.1-10 µM of IN-2001. In MCF-7 cells, cyclin D1 levels were suppressed by 84.1%, 56.1%, and 40.7% at 0.1, 0.5, and 1 µM IN-2001, respectively. Taken together, these results demonstrate that specific changes in the expression of cell cycle-related genes may lead to cell cycle arrest, ultimately resulting in growth inhibition via upregulation of p21WAF1, and downregulation of cyclin D1 in human breast cancer cells.
DISCUSSION
In this study, we reported that IN-2001 HDAC inhibitor effectively inhibited the proliferation of human breast cancer cells. Previous data, published on MCF-7 and MDA-MB-231 cells using TSA or sodium butyrate as an inhibitor of HDAC, also supported a higher sensitivity of ER-positive breast cancer cells [18]. This differential response was not associated with a modification of drug efflux via the multidrug resistance system or with a global modification of HAT/HDAC activities [18-24]. They suggested that p21WAF1 gene expression, in relation with ERα, could play a role in this differential response of breast cancer cells to hyperacetylating agents. In ER-positive breast cancer cells, the p21WAF1 gene was more sensitive to TSA regulation and was expressed at higher levels. Moreover, the re-expression of ERα following adenoviral infection of ER-negative breast cancer cells increased both growth inhibitory activity of TSA and p21WAF1 protein accumulation. In the current study, we found that HDAC inhibitors induced the expression of CDK inhibitor, such as p21WAF1 and the induction of p21WAF1 was highly correlated with the growth inhibitory effect of HDAC inhibitors. Further studies are needed to clarify the implications of ER status in the regulation of p21WAF1 by HDAC inhibitors.
HDAC inhibitors have been reported to induce apoptosis in various human cancer cell lines, such as MCF-7 breast cancer [25], small lung cancer [26], HOS osteosarcoma [27], and HCT116 colon cancer [28]. Ruefli et al. [29] and Rosato et al. [30] suggested that HDAC inhibitor-induced apoptosis involved two different mechanisms, the intrinsic pathway and the extrinsic pathway. The relative importance of these two pathways depends on the cell type and class of HDAC inhibitors used. In previous study, HDAC inhibitors effectively induced apoptosis in a dose-dependent manner, as visualized by FACS analysis. At the same time, HDAC inhibitors activated caspase 3/7 and increased cytochrome C release, which was accompanied by the increase of the relative ratio of Bax/Bcl-2 [31-33]. These findings indicate that HDAC inhibitor may induce caspase-dependent apoptosis partially through the mitochondrial pathway, accompanied by an increased ratio of Bax/Bcl-2 and cytochrome C release. In particular, MDA-MB-231 cells treated with HDAC inhibitor showed strong activation of the intrinsic pathway. The implication of the extrinsic pathway in HDAC inhibitor-induced apoptosis had not been determined in this study. To make this clear, further studies are required, including examination on the death receptor activity and the initiator caspases (e.g., caspase 8 and caspase 9) activation. In addition, HDAC inhibitors increased ROS induction. In particular, remarkable induction of ROS generation was observed with TSA and LAQ, which showed potent growth inhibitory effects and inducibility of apoptosis [34]. These data are compatible with the notion that HDAC inhibitors are potent inducers of ROS and that induced ROS are important in the HDAC inhibitor-mediated apoptosis.
Considering the role of p21WAF1 as a negative regulator of cell cycle progression, our observation that HDAC inhibitors commonly induced p21WAF1 expression both in vivo and in vitro breast cancer model, designating a possible anti-tumor mechanism of HDAC inhibitors. HDAC inhibitors upregulate the expression of p21WAF1 and increased p21WAF1 may trigger cell cycle arrest, resulting in inhibition of cell proliferation. In summary, IN-2001 HDAC inhibitor inhibited human breast cancer MCF-7 cell proliferation via cell cycle arrest at G2/M arrest as well as an increase in apoptosis.